
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspective:
September 2025
View 2025 - 2021 Archive || 2020 Archive || 2019 Archive ||
2018 Archive || 2017 and Earlier Archive
April 2023:
March 2023:
February 2023:
Alpha-Synuclein Abundance and Localization are Regulated by the RNA-Binding Protein PUMILIO1
The Role of Astrocytes in Human Huntington's Disease Pathology
Early Proteasome Downregulation and Dysfunction Drive Proteostasis Failure in Alzheimer's Disease
The Role of Alpha-Synuclein in Synucleinopathy: Impact on Lipid Regulation at Mitochondria-ER
Associations Between Hearing Loss and Dementia in a Large Electronic Health Record System
APOE ε4-Associated Heterogeneity of Neuroimaging Biomarkers Across the Alzheimer's Disease Continuum
APOE and Alzheimer's Disease and Related Dementias Risk Among 12,221 Hispanics/Latinos
A Human Brain Map of Mitochondrial Respiratory Capacity and Diversity
ANXA11 Biomolecular Condensates Facilitate Protein-Lipid Phase Coupling on Lysosomal Membranes
Axonal Transport of CHMP2b Is Regulated by Kinesin-Binding Protein and Disrupted by CHMP2bintron5
Sleep Genetics and Cognitive Changes over Time: The Moderating Effect of Age and the Role of Brain
Emerging Roles for Tubulin PTMs in Neuronal Function and Neurodegenerative Disease
Inflammatory Biomarkers Profiles and Cognition Among Older Adults
Synaptic and Cognitive Impairment Associated with L444P Heterozygous Glucocerebrosidase Mutation
Regulation of Synapse Density by Pumilio RNA-Binding Proteins
CD33 and SHP-1/PTPN6 Interaction in Alzheimer's Disease
Cellular Communities Reveal Trajectories of Brain Ageing and Alzheimer's Disease
Epigenetic and Genetic Risk of Alzheimer Disease from Autopsied Brains in two Ethnic Groups
Multi-Omic Analysis of Huntington's Disease Reveals a Compensatory Astrocyte State
Design and Methods of the Early Age-Related Hearing Loss Investigation Randomized Controlled Trial
Updated Safety Results From Phase 3 Lecanemab Study in Early Alzheimer's Disease
The Broken Alzheimer's Disease Genome
Rare Genetic Variation in Fibronectin 1 (FN1) Protects Against APOEε4 in Alzheimer's Disease
Cell Subtype-Specific Effects of Genetic Variation in the Alzheimer's Disease Brain
Diet, Pace of Biological Aging, and Risk of Dementia in the Framingham Heart Study
A Comparative Study of Structural Variant Calling in WGS from Alzheimer's Disease Families
Glucocorticoid Stress Hormones Stimulate Vesicle-Free Tau Secretion and Spreading in the Braint
The Effects of Insufficient Sleep and Adequate Sleep on Cognitive Function in Healthy Adults
ZCCHC17 Modulates Neuronal RNA Splicing and Supports Cognitive Resilience in Alzheimer's Disease
Effects of Lithium on Serum Brain-Derived Neurotrophic Factor in Alzheimer's Patients with Agitation
2023 Taub Institute Grants for Emerging Research (TIGER) Awardees!
Rie1 and Sgn1 Form an RNA-Binding Complex that Enforces the Meiotic Entry Cell Fate Decision
Memory and Language Cognitive Data Harmonization Across the United States and Mexico
Education as a Moderator of Help Seeking Behavior in Subjective Cognitive Decline
Multicellular Communities are Perturbed in the Aging Human Brain and Alzheimer's Disease
The Neuropathological Landscape of Hispanic and non-Hispanic White Decedents with Alzheimer Disease
The Early-Onset Alzheimer's Disease Whole-Genome Sequencing Project: Study Design and Methodology
Polygenic Risk Score Penetrance & Recurrence Risk in Familial Alzheimer Disease
High School Quality is Associated with Cognition 58 Years Later
Glucocorticoid-Driven Mitochondrial Damage Stimulates Tau Pathology
A Global View of the Genetic Basis of Alzheimer Disease
ARIA in Patients Treated with Lecanemab (BAN2401) in a Phase 2 Study in Early Alzheimer's Disease
Microglia Reactivity Entails Microtubule Remodeling from Acentrosomal to Centrosomal Arrays
Genuine Selective Caspase-2 Inhibition with new Irreversible Small Peptidomimetics
Cell Type-Specific Changes Identified by Single-Cell Transcriptomics in Alzheimer's Disease
Brain Aging Among Racially and Ethnically Diverse Middle-Aged and Older Adults
First Place: Neuroproteasome Localization and Dysfunction Modulate Pathology in Alzheimer's Disease
Clearance of an Amyloid-Like Translational Repressor is Governed by 14-3-3 Proteins
Diet Moderates the Effect of Resting State Functional Connectivity on Cognitive Function
Retromer Deficiency in Tauopathy Models Enhances the Truncation and Toxicity of Tau
Progranulin Mutations in Clinical and Neuropathological Alzheimer's Disease
Wolframin is a Novel Regulator of Tau Pathology and Neurodegeneration
Homotypic Fibrillization of TMEM106B Across Diverse Neurodegenerative Diseases
Correlation of Plasma and Neuroimaging Biomarkers in Alzheimer's Disease
Tubulin Tyrosination Regulates Synaptic Function and is Disrupted in Alzheimer's Disease
The Penalty of Stress - Epichaperomes Negatively Reshaping the Brain in Neurodegenerative Disorders
The Neuronal Retromer can Regulate Both Neuronal and Microglial Phenotypes of Alzheimer's Disease
Deep Learning Improves Utility of Tau PET in the Study of Alzheimer's Disease
Age of Onset of Huntington's Disease in Carriers of Reduced Penetrance Alleles
Caspase-9: A Multimodal Therapeutic Target With Diverse Cellular Expression in Human Disease
Midlife Vascular Factors and Prevalence of Mild Cognitive Impairment in Late-Life in Mexico
The Association Between Sex and Risk of Alzheimer's Disease in Adults with Down Syndrome
Marked Mild Cognitive Deficits in Humanized Mouse Model of Alzheimer's-Type Tau Pathology
Rapid ATF4 Depletion Resets Synaptic Responsiveness after cLTP
Polygenic Risk Score for Alzheimer's Disease in Caribbean Hispanics
Recognition Memory and Divergent Cognitive Profiles in Prodromal Genetic Frontotemporal Dementia
The Microtubule Cytoskeleton at the Synapse & The Synaptic Life of Microtubules
Optimizing Subjective Cognitive Decline to Detect Early Cognitive Dysfunction
The AD Tau Core Spontaneously Self-Assembles and Recruits Full-Length Tau to Filaments
Olfactory Impairment is Related to Tau Pathology and Neuroinflammation in Alzheimer's Disease
Pathogenic Role of Delta 2 Tubulin in Bortezomib-Induced Peripheral Neuropathy
1: Frequency of Microvascular Pathology and Hippocampal Atrophy on Magnetic Resonance Imaging in a Community Study of Alzheimer's Disease with Blood-Based Biomarkers
2: The Spatial Landscape of Glial Pathology and T Cell Response in Parkinson's Disease Substantia Nigra
3: Rare FN1 Missense Mutations Indicate a Protective Role Against Lewy Body Dementia in APOEε4 Homozygous Carriers
2: The Spatial Landscape of Glial Pathology and T Cell Response in Parkinson's Disease Substantia Nigra
3: Rare FN1 Missense Mutations Indicate a Protective Role Against Lewy Body Dementia in APOEε4 Homozygous Carriers
 |  |  | ||
| Tamil Iniyan Gunasekaran, PhD | Adam M. Brickman, PhD | Richard Mayeux, MD, MSc |
Alzheimer’s disease (AD) often coexists with cerebrovascular disease, yet the relationship between these overlapping brain pathologies is still not well defined. In the current study, we set out to understand how cerebrovascular disease interacts with blood-based biomarkers of AD in a multiethnic, community-based cohort from the Washington Heights-Hamilton Heights-Inwood Columbia Aging Project (WHICAP). In 685 participants representing European, African American, and Caribbean Hispanic ancestry, our team measured blood biomarkers of AD alongside MRI markers of cerebrovascular disease and neurodegeneration. Participants were categorized into biomarker-negative controls (BM-CTL), biomarker-positive individuals without dementia (preclinical AD; BM+CTL), biomarker-positive individuals with AD dementia (BM+AD), and individuals with other dementias (BM-Dem), based on established thresholds for P-tau181 and P-tau217.
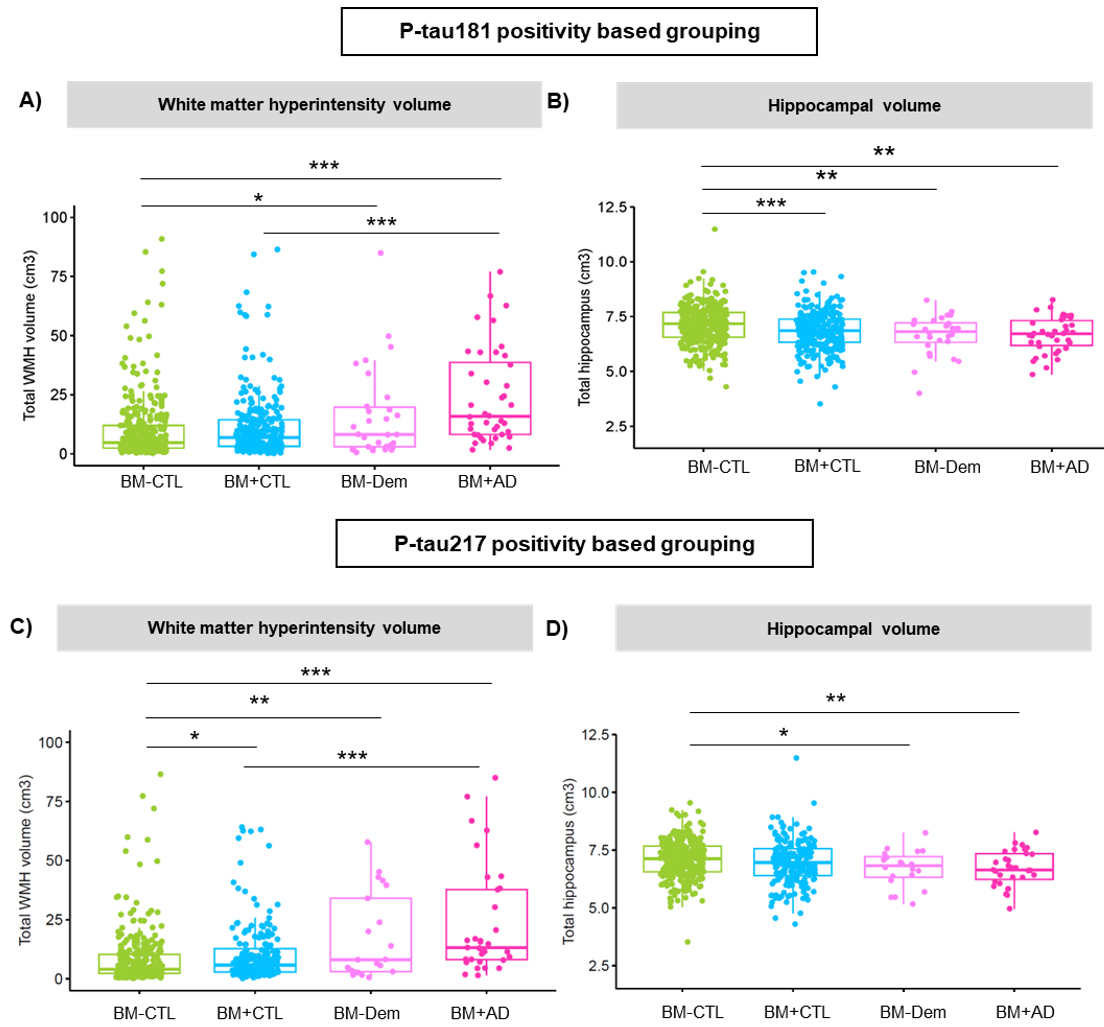
Figure 3. Comparison of neuroimaging measures across clinical groups defined by P-tau181 (top panels) and P-tau217 (bottom panels). White matter hyperintensity volume is shown on the left (A, C) and hippocampal volume on the right (B, D). Group differences are indicated with significance levels (*p < 0.05; **p < 0.01; **p < 0.001).
As recently reported in Annals of Neurology, we found that biomarker-positive individuals including those without overt cognitive symptoms had greater white matter hyperintensity (WMH) burden, more silent brain infarcts, and smaller hippocampal volumes than biomarker-negative controls. Interestingly, the two tau biomarkers showed different associations: P-tau217 was more strongly linked to WMH burden, while P-tau181 was more closely tied to hippocampal atrophy. These patterns were evident even in those without dementia, suggesting that vascular changes may emerge early in the disease process, alongside subtle neurodegeneration.
Our findings highlight that cerebrovascular disease is not simply a co-occurring condition but a frequent and important contributor to AD pathology. They also underscore the value of integrating blood biomarkers with neuroimaging to capture the complexity of AD, particularly in aging, multiethnic populations such as WHICAP.
Tamil Iniyan Gunasekaran, PhD
Postdoctoral Research Scientist in the Gertrude H. Sergievsky Center
tg2803@cumc.columbia.edu
Adam Brickman, PhD
Professor of Neuropsychology (in Neurology, the Taub Institute and the Gertrude H. Sergievsky Center)
amb2139@cumc.columbia.edu
Richard Mayeux, MD, MSc
Gertrude H. Sergievsky Professor of Neurology, Psychiatry and Epidemiology (in the Taub Institute and the Gertrude H. Sergievsky Center)
rpm2@cumc.columbia.edu
The Spatial Landscape of Glial Pathology and T Cell Response in Parkinson's Disease Substantia Nigra

Osama Al-Dalahmah, MD, PhD
In our recent study published in Nature Communications, we sought to create a detailed map of the immune and glial cell landscape within the brains of patients with Parkinson’s disease (PD). For decades, mounting evidence has suggested that the immune system plays a role in PD, but the specific characteristics of this response within the most affected brain regions have remained unclear. We focused on the substantia nigra, the movement-control hub that degenerates in PD, and discovered that it is infiltrated by CD8+ T cells. Using T cell receptor sequencing and single-nucleus RNAseq (snRNAseq), we found that these T cells were clonally expanded, indicating they were mounting a targeted response to a specific trigger. Excitingly, we found that the receptors on these brain T cells had strong similarities to those known to react to α-synuclein, the protein that aggregates in PD, providing a crucial link between the disease's primary pathology and the resulting immune reaction. Our snRNAseq and immunohistochemical studies allowed us to further characterize the T cells in the substantia nigra, and we found that T cells in the substantia nigra assume a resident-memory state, further supporting that they are antigen-experienced.
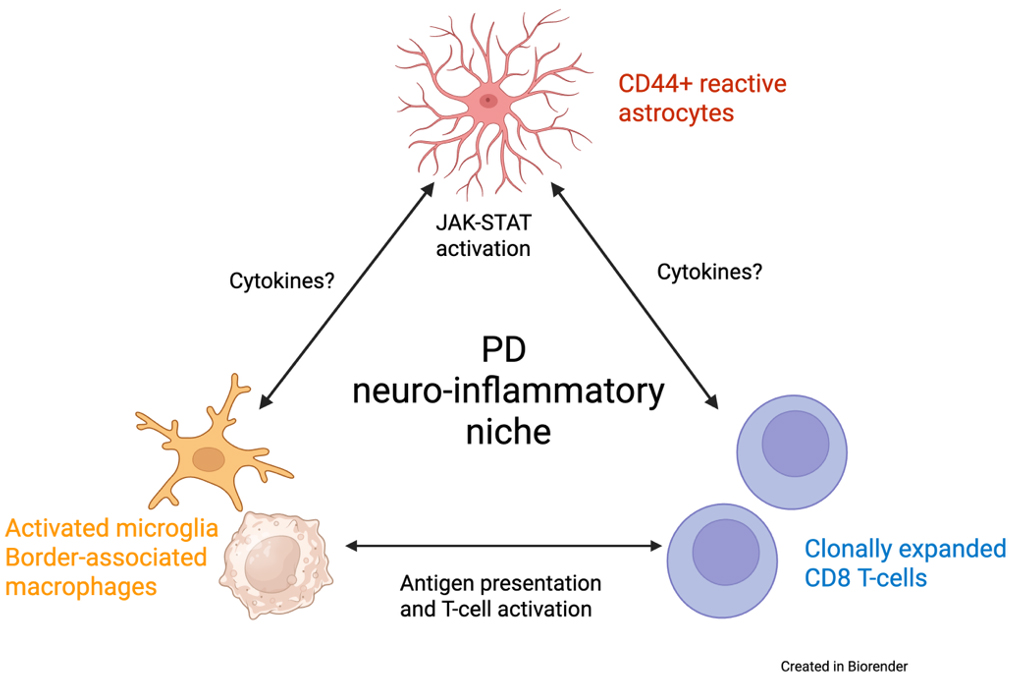
Graphical abstract.
Our investigation went a step further by using spatial transcriptomics and validation immunohistochemical studies to understand how these T cells interact with their environment. We discovered that T cells in the PD brain exist within a "neuro-inflammatory niche" – see schematic, in close proximity to activated microglia and a particular type of reactive astrocyte that expresses high levels of the surface protein CD44. These CD44+ astrocytes were significantly increased in the PD substantia nigra and were associated with elevated neuroinflammatory signaling pathways, such as the JAK/STAT pathway. We pursued this finding by interfering with CD44 expression in cultured astrocytes using a gene knockdown approach. The results showed that CD44 knockdown reduced the gene expression changes associated with several activated neuro-inflammatory pathways, including JAK-STAT, nominating CD44 as a potential therapeutic target.
In summary, our study provided a detailed, single-cell, and spatially localized map of the neurodegenerative niche in PD. Furthermore, our findings provide a direct link between a reactive astrocyte state characterized by an elevated CD44 expression and neurodegeneration, as we have observed in Huntington’s disease, as well as neuroinflammation, given the involvement of clonally expanded T cells in the pathology. This result is particularly promising because it moves beyond mere observation and identifies CD44 as a potential therapeutic target, not only in PD, but also in other neurodegenerative diseases. We now have NIH funding to pursue this target in Alzheimer’s disease. By blocking CD44, we might be able to interrupt the vicious cycle of neuroinflammation, offering a new strategy to slow the progression of neurodegenerative diseases.
Osama Al-Dalahmah, MD, PhD
Assistant Professor in Pathology and Cell Biology
oa2298@cumc.columbia.edu

Badri N. Vardarajan, PhD, MS
The APOE ε4 allele is the strongest genetic risk factor for late-onset Alzheimer's disease, with ε4 homozygotes showing nearly complete disease penetrance, yet some elderly ε4 carriers remain cognitively healthy, suggesting protective genetic factors may exist. Previously, in collaboration with Drs. Kizil and Mayeux, we investigated modifying genetic factors that confer resilience to high risk elderly ε4 carriers through whole genome sequencing of 3,500 individuals from multiplex families. We identified a rare Fibronectin-1 (FN1) missense variant that segregated with cognitively healthy elderly APOE ε4 carriers and reduced Alzheimer's risk by 71% while delaying disease onset by four years in an independent cohort. Using autopsied brain tissue and animal models, we demonstrated that this variant protects against Alzheimer's disease by preventing excess fibronectin accumulation at the blood-brain barrier, enhancing amyloid clearance and suggesting new therapeutic strategies targeting fibronectin regulation.
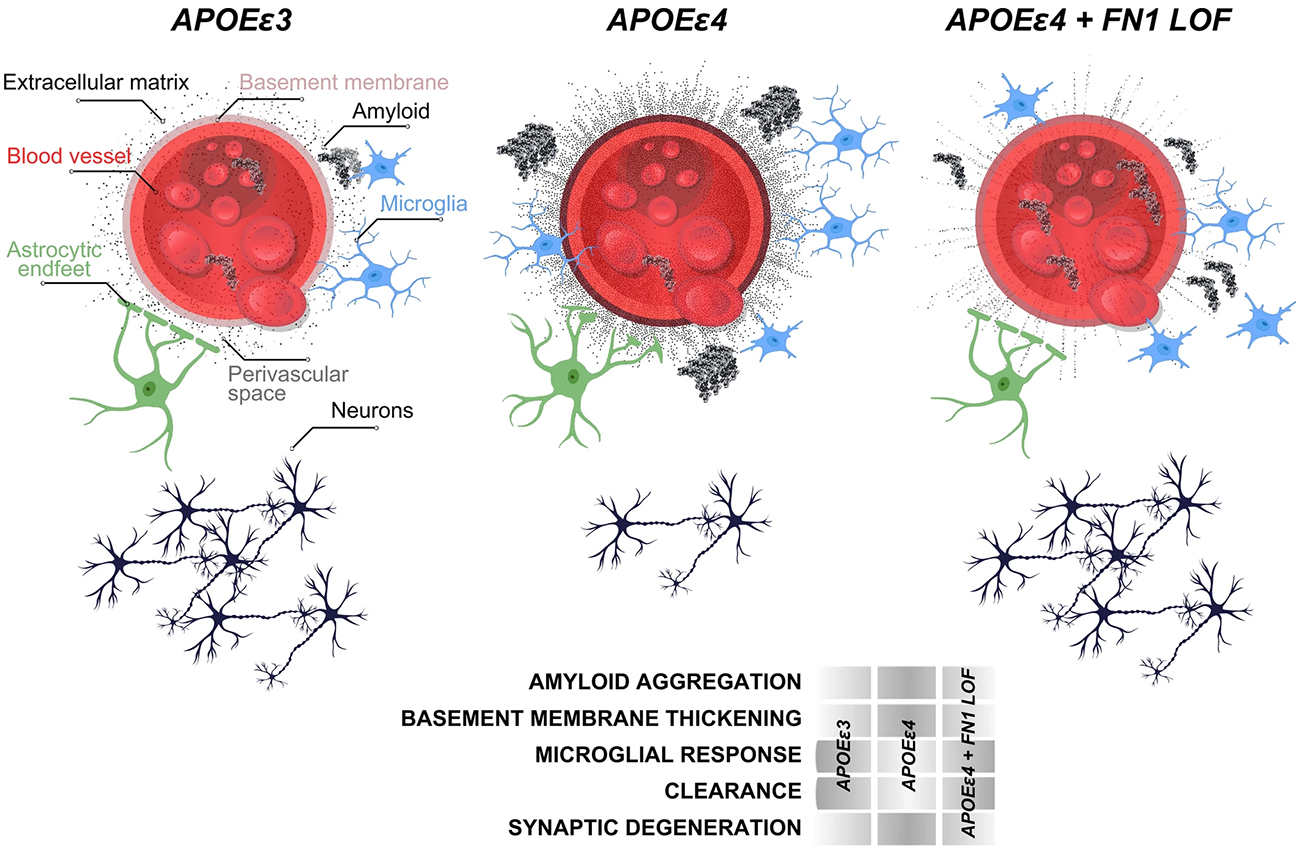
Figure. Schematic abstract for the protective effect of FN1 variants.
In this study, we investigated the role of FN1 mutations in Lewy body dementia, to build on our previous findings. In collaboration with investigators from Columbia and the NINDS, we extracted a carefully curated cohort of 212 LBD cases and 61 neurologically healthy controls who were all APOE ε4 homozygotes from European ancestry populations. What made this analysis particularly rigorous was our focus on pathologically definite cases—nearly 80% of our LBD patients had been confirmed through autopsy, giving us confidence that we were studying true disease. We used advanced statistical methods to examine rare FN1 missense and loss-of-function variants while carefully controlling for age, sex, and population structure.
As recently reported in Acta Neuropathologica, we identified five different FN1 missense mutations, but their distribution told a compelling story: these variants appeared in 8.6% of healthy controls but only 1.4% of LBD patients, yielding a statistically significant p-value of 0.008 for protection against disease. When we expanded our analysis beyond APOE ε4 homozygotes to include heterozygotes and non-carriers, we found an even stronger protective signal in APOE ε4 non-carriers, with a p-value of 3.32 × 10⁻⁶. None of the FN1 mutation carriers had variants in known LBD risk genes like GBA or SNCA, suggesting that FN1's protective effect operates through an independent mechanism. All three LBD cases carrying FN1 mutations had pathologically confirmed disease, with two showing Alzheimer's co-pathology, reinforcing the complex interplay between different neurodegenerative processes.
While we acknowledge that our relatively small sample size represents a limitation, we believe our findings provide important corroborating evidence that FN1 missense mutations confer protection not just against Alzheimer's disease, but also against Lewy body dementia. This protective effect appears to extend beyond APOE ε4 homozygotes to other APOE genotypes, suggesting a broader biological mechanism at play. This work demonstrates the value of examining shared genetic architecture across different neurodegenerative diseases and opens new avenues for understanding how fibronectin-mediated pathways might be leveraged for therapeutic intervention in multiple forms of dementia.
Badri N. Vardarajan, PhD, MS
Associate Professor of Neurological Science (in Neurology and the Gertrude H. Sergievsky Center and the Taub Institute)
bnv2103@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.