
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspective:
April 2023
1: Glucocorticoid-Driven Mitochondrial Damage Stimulates Tau Pathology
2: A Global View of the Genetic Basis of Alzheimer Disease
3: ARIA in Patients Treated with Lecanemab (BAN2401) in a Phase 2 Study in Early Alzheimer's Disease
2: A Global View of the Genetic Basis of Alzheimer Disease
3: ARIA in Patients Treated with Lecanemab (BAN2401) in a Phase 2 Study in Early Alzheimer's Disease
Glucocorticoid-Driven Mitochondrial Damage Stimulates Tau Pathology
 |  |  | ||
| Fang Du, PhD | Qing Yu, PhD | Clarissa Waites, PhD |
Chronic stress is a known risk factor for depression and Alzheimer’s disease (AD). Stress and glucocorticoids, the hormones released during stressful experiences, damage the brain in multiple ways, leading to neuron and synapse loss, mood dysregulation, and memory impairment. Two major drivers of glucocorticoid-induced brain damage are mitochondrial dysfunction, causing widespread disruption of cellular energy metabolism, and Tau pathology (i.e. hyperphosphorylation/oligomerization of Tau protein), causing disruption of neuronal functions including synaptic transmission, axon transport, and protein clearance. How glucocorticoids induce mitochondrial dysfunction and Tau pathology at the cellular/molecular level, and how these two events are causally related, remains unclear.

Graphical Abstract
In Du et al., now published online in BRAIN, we investigated these questions with experiments in cultured neurons treated with glucocorticoids and mice administered glucocorticoids to mimic stress. At the cellular level, we found that glucocorticoids promote mitochondrial dysfunction by stimulating the opening of a channel on the inner mitochondrial membrane called the ‘mitochondrial permeability transition pore’ (mPTP). Opening of the mPTP compromises mitochondrial function, decreasing production of ATP for cellular energy needs while increasing production of reactive oxygen species (ROS), which stimulates further mitochondrial damage and cellular toxicity. Mechanistically, we showed that glucocorticoids induce mPTP opening by promoting transcription of the mPTP activating component, Cyclophilin D. Interestingly, we found that inhibition of Cyclophilin D via either genetic or pharmacological means was not only protective against glucocorticoid-induced mitochondrial damage, but also against Tau hyperphosphorylation and oligomerization in cultured neurons. These findings illuminate a key mechanism by which glucocorticoids cause mitochondrial damage in neurons and suggest that Tau pathology occurs downstream of this damage.
Since our in vitro experiments indicated that mPTP inhibition prevents glucocorticoid-induced mitochondrial dysfunction and Tau pathology in cultured neurons, we next looked for a therapeutic approach to inhibit mPTP opening in vivo. To this end, we tested the chemical compound mito-apocynin, a mitochondrially-targeted inhibitor of ROS production that is orally administered and protective against mitochondria-related toxicity in several mouse models. We found that mito-apocynin is indeed able to prevent glucocorticoid-induced mPTP opening and mitochondrial damage. Moreover, this compound also protects against glucocorticoid-induced Tau pathology, synapse loss, and behavior deficits (learning/memory impairment, anxiety, depressive behavior) in mice, indicative of its protection against stress-related brain damage.
We then tested the efficacy of mito-apocynin in cytoplasmic hybrid (cybrid) cells, an ex vivo AD model wherein endogenous mitochondria are replaced with mitochondria from AD subjects. Compared to cybrid cells containing mitochondria from control subjects, AD cybrids exhibit dramatically higher levels of mitochondrial ROS and hyperphosphorylated/oligomerized Tau. These phenotypes were restored to the level of control cybrids by mito-apocynin, demonstrating the compound’s promise for protecting against mitochondrial damage and Tau pathology induced by AD. Finally, we found that the glucocorticoid receptor antagonist mifepristone, which blocks glucocorticoid signaling, is similarly able to rescue these phenotypes in AD cybrid cells. Together, our findings link glucocorticoids to mitochondrial dysfunction and Tau pathology in the context of Alzheimer’s disease and suggest that mitochondria are promising therapeutic targets for mitigating stress- and Tau-related brain damage.
Clarissa Waites, PhD
Associate Professor of Pathology and Cell Biology and Neuroscience (in the Taub Institute)
cw2622@cumc.columbia.edu
A Global View of the Genetic Basis of Alzheimer Disease
 |  | |
| Christiane Reitz, MD, PhD | Richard Mayeux, MD, MSc |
Large-scale genome-wide analyses indicate that 70 or more genes or loci contribute to Alzheimer’s Disease (AD). However, a major factor limiting progress is that most genetic data have been obtained from non-Hispanic white individuals in Europe and North America. From a global perspective, the human migration events that have taken place over thousands of years of human evolutionary history have shaped the genetic diversity among current populations and have resulted in substantial differences in allele frequencies and the extent to which common alleles are inherited together (linkage disequilibrium). Anatomically modern humans originated in Africa, and the population subsequently expanded outwards. The population remaining in Africa maintained a larger effective size and had the most time for genetic recombination than other populations. As a result, individuals with a greater degree of African ancestry have less linkage disequilibrium and higher genetic diversity than individuals with a smaller degree of African ancestry. Populations outside Africa show less genetic diversity with a linear decline of heterozygosity and flattening of the ancestral allele frequency spectrum, as a function of geographic distance from Africa.
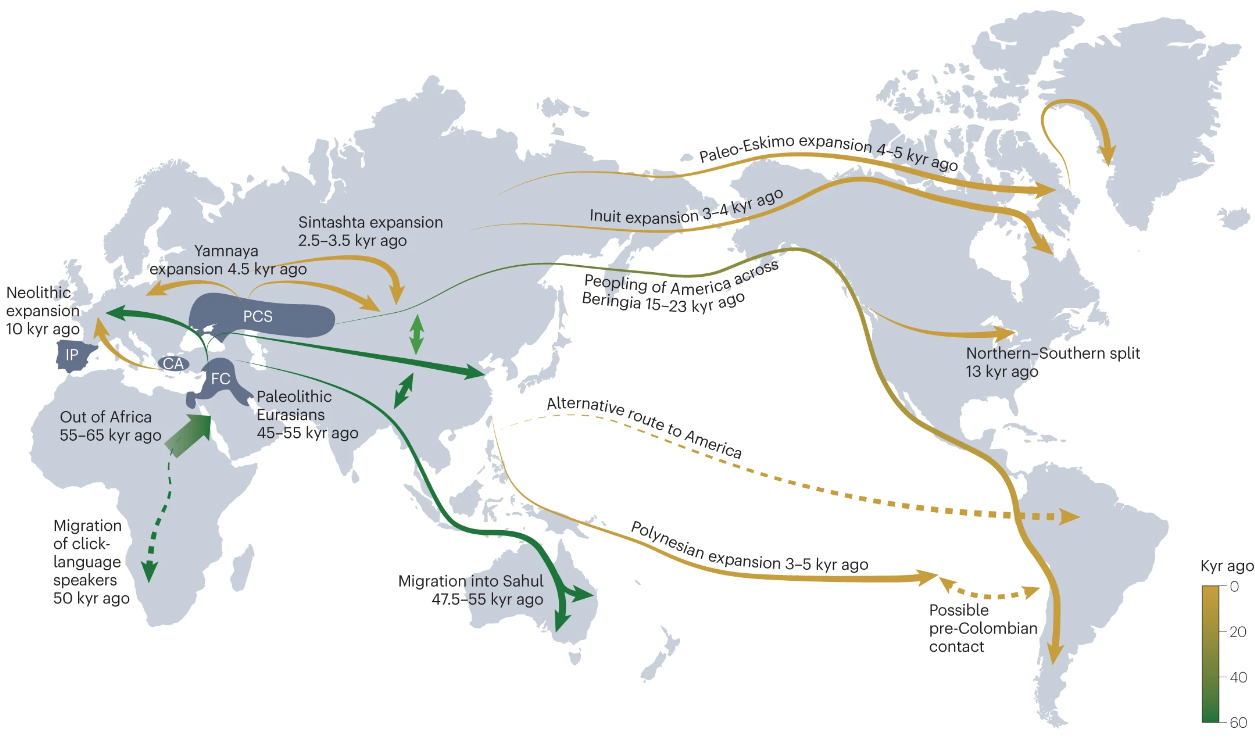 Figure 2. Timeline and routes of human migration inferred from genomic data. Dashed lines represent routes of migration that remain controversial. CA, Central Anatolia; FC, Fertile Crescent; IP, Iberian Peninsula; kyr, thousand years; PCS, Pontic-Caspian steppe. Adapted from ref. 208, Springer Nature Limited.
|
These ancestral differences have profound effects on the associations of genetic variants with traits and disease in different populations. Although some associations between genetic markers and disease are present across populations — providing important evidence for a shared genetic basis of disease risk — genetic risk prediction models developed from one ancestral group do not perform as well when applied to other ancestral groups. In an individual, overall genetic disease risk is determined by multiple genetic variants from a combination of ancestral backgrounds. For admixed groups such as Latinx, it is therefore essential to understand AD risk in indigenous African and Amerindian individuals. The subset of human genetic variation left behind in Africa can only be studied in individuals with a high degree of African ancestry.
Fortunately, emerging genetic data from other regions —including Africa, Asia, India and South America — are now providing information on the disease from a broader range of ethnicities. In our recent paper published in Nature Reviews Neurology, we review the current knowledge on AD genetics in populations across the world. We summarize novel large-scale genomic efforts in undersampled populations specifically designed to identify a broader range of genetic variations that influence AD, and outline gaps that need to be addressed to achieve a complete picture of the genetic and molecular factors that drive AD in individuals across the globe. The aim is to move development of personalized approaches to AD in individuals of other ethnicities forward.
Christiane Reitz, MD, PhD
Associate Professor of Neurology and Epidemiology (in the Taub Institute and the Gertrude H. Sergievsky Center)
cr2101@cumc.columbia.edu
Richard Mayeux, MD, MSc
Gertrude H. Sergievsky Professor of Neurology, Psychiatry and Epidemiology (in the Taub Institute and the Gertrude H. Sergievsky Center)
rpm2@cumc.columbia.edu

Lawrence S. Honig, MD, PhD
ARIA in Patients Treated with Lecanemab (BAN2401) in a Phase 2 Study in Early Alzheimer's Disease
Over the past decade, clinical investigators from the Taub Institute and elsewhere have been involved in the development of immunotherapies—both active immunizations and passive immunization antibody therapies—including bapineuzumab, crenezumab, solanezumab, gantenerumab, and lecanemab, aimed at lowering cerebral amyloid beta burden in patients with Alzheimer's disease (AD). We have observed amyloid-related imaging abnormalities (ARIA) on brain imaging after treatment with some anti-amyloid immunotherapies, yet the exact pathophysiology underlying ARIA remains uncertain. In a study recently published in Alzheimer’s & Dementia: Translational Research & Clinical Interventions, we present detailed results on the ARIA profile of lecanemab treatment from a large phase 2 (Study 201) core trial in early AD and open-label extension (OLE) phases.
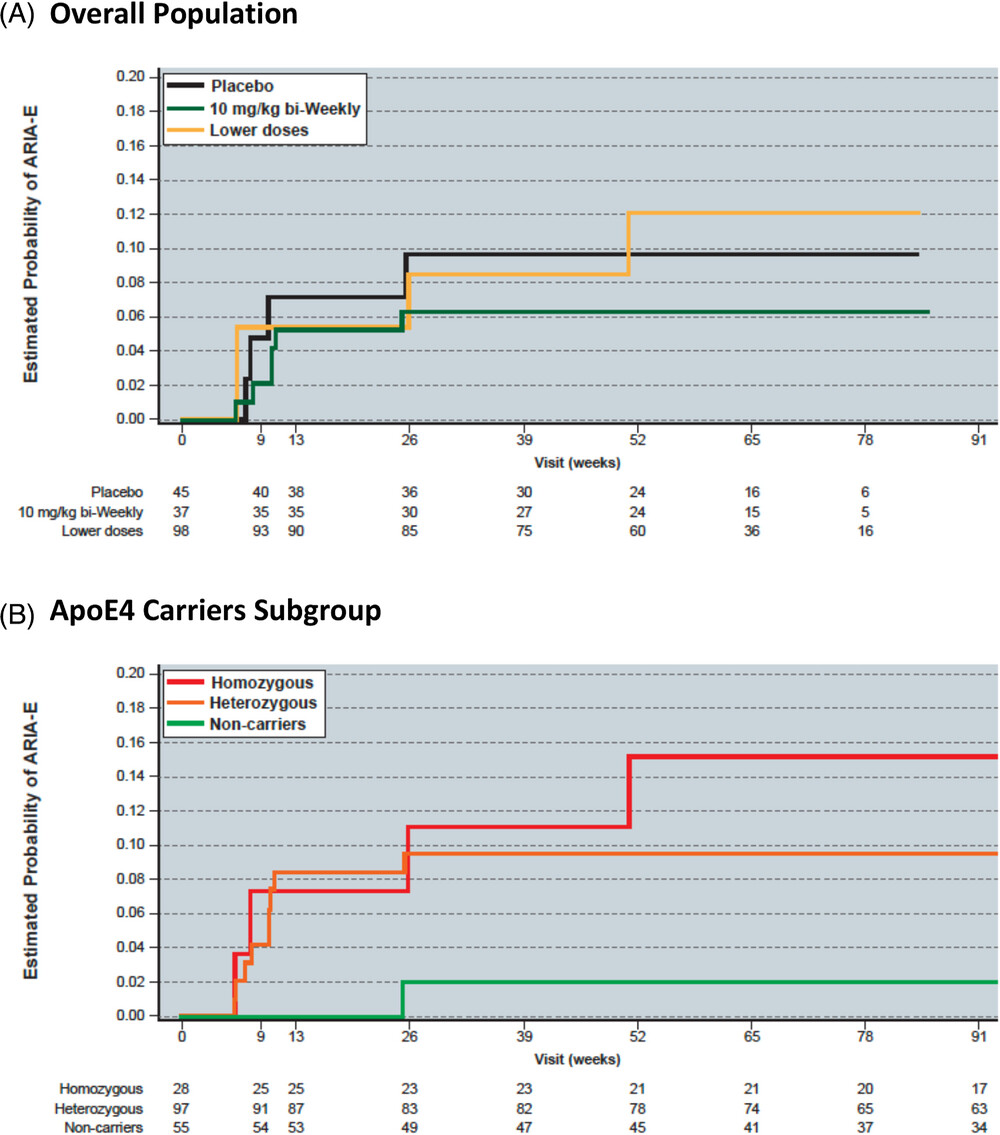
Figure 2. Kaplan-Meier estimate of ARIA-E risk in subjects receiving 10 mg/kg biweekly lecanemab in the Study 201 OLE for the (A) overall ole population and the (B) ApoE4 carriers subgroup.
Together with Marwan Sabbagh (University of Arizona College of Medicine-Phoenix) and colleagues at Eisai, Inc., we found that the incidence of observed ARIA-E cases was consistent in the core and OLE phases for the 10 mg/kg biweekly dose. Observed ARIA in the Study 201 and OLE was largely asymptomatic, and radiographically mild to moderate; it generally occurred within the first 3 months of treatment, and occurred more commonly in persons who carry one or two apolipoprotein E4 alleles. Importantly, asymptomatic ARIA-E can be dosed through in some mild-to-moderate cases. Overall, our findings support use of lecanemab therapy at 10 mg/kg biweekly without requiring titration of the dosage.
Further evaluation of lecanemab in the recently completed phase 3 Clarity AD study in early Alzheimer's disease appears to show similar results. Research on a lecanemab subcutaneous formulation is ongoing and has additional potential for reduced incidence of ARIA-E versus the IV formulation, due to a lower peak blood concentration of the drug.
Lawrence S. Honig, MD, PhD
Professor of Neurology (in the Taub Institute and the Gertrude H. Sergievsky Center) at CUMC
lh456@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.