
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspectives:
Best Poster Presentations
Taub Institute Retreat November 2023

Natalie Edwards
PhD Student
Cerebrovascular Disease Drives Alzheimer Plasma Biomarker Concentrations in Adults with Down Syndrome
Natalie C. Edwards1,2,3, Patrick J. Lao1,2, Mohamad J. Alshikho1,2, Olivia M. Ericsson1,2, Batool Rizvi4, Lisi Flores Aguilar5, Melissa E. Petersen6, Sid O’Bryant6, Dana L. Tudorascu7, José Gutierrez2, Donna M. Wilcock8,9,10, Elizabeth Head5, & Adam M. Brickman1,2
1) Taub Institute for Research on Alzheimer’s Disease and the Aging Brain, Columbia University, New York City, NY, USA.
2) Department of Neurology, Vagelos College of Physicians and Surgeons, Columbia University, New York City, NY, USA.
3) Department of Neuroscience, Columbia University, New York City, NY, USA. 4) Department of Neurobiology & Behavior, University of California, Irvine, CA, USA.
4) Department of Neurobiology & Behavior, University of California, Irvine, CA, USA.
5) Department of Pathology and Laboratory Medicine, University of California Irvine School of Medicine, University of California, Irvine, CA, USA.
6) University of North Texas Health Science Center, Fort Worth, TX, USA.
7) Department of Psychiatry, University of Pittsburgh, Pittsburgh, PA, USA.
8) Stark Neurosciences Research Institute, Indiana University School of Medicine, Indianapolis, IN, USA.
9) Department of Neurology, Indiana University School of Medicine, Indianapolis, IN, USA.
10) Department of Anatomy, Cell Biology and Physiology, Indiana University School of Medicine, Indianapolis, IN, USA.
By age 40 years, adults with Down syndrome (DS) develop Alzheimer’s disease (AD) neuropathology and ~90% progress to dementia in their 50’s. Despite having few systemic vascular risk factors, individuals with DS have significant cerebrovascular disease that increases in severity with the clinical progression of AD, suggesting a vascular component of DS-AD that is not solely attributable to vascular risk factors. In the current study, we examined pathways that may link cerebrovascular disease to AD pathophysiology in adults with DS by testing the association among markers of cerebrovascular disease, inflammation/astrocytosis, and blood-based AD biomarkers across clinical AD diagnostic groups (i.e., cognitively stable, mild cognitive impairment [MCI], and clinical AD).
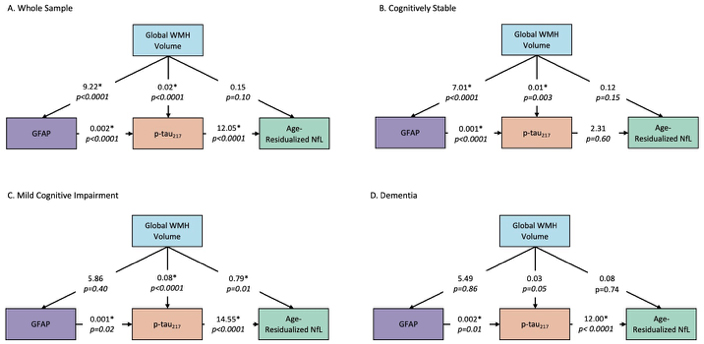
Path models for biomarker progression across diagnostic groups. Statistical modeling calculates relative causal relationships among different pathophysiological contributors. Larger numbers (regression coefficients) signify stronger direct effects.
One hundred eighty-five participants from the Alzheimer’s Biomarkers Consortium – Down Syndrome (mean age[SD]=45.2[9.3] years) with available MRI and plasma biomarker data were included in the study. Whole-brain white matter hyperintensity (WMH) volumes were derived from T2-weighted FLAIR MRI scans as a marker of small vessel cerebrovascular disease, and plasma Aβ42/Aβ40, p-tau217, glial fibrillary acidic protein (GFAP), and neurofilament light (NfL) concentrations were measured with SIMOA assays, reflecting the amyloid, tau, inflammatory, and neurodegenerative pathophysiology, respectively implicated in AD. After examining bivariate relationships between WMH volume and plasma biomarker concentrations using Pearson correlation tests, we conducted a series of path analyses to examine potential causal pathways that may link cerebrovascular disease with AD pathophysiology in the total sample and in groups stratified by clinical diagnosis.
Larger WMH volume was associated with higher p-tau217, GFAP, and NfL, but not with Aβ concentrations. Path analyses suggest that among individuals with DS, cerebrovascular disease promotes AD-related neurodegeneration by increasing inflammation/astrocytosis and tau pathophysiology, particularly in presymptomatic and early phases of AD. Our findings show codependency and suggest a causal relationship between cerebrovascular disease and the progression of AD pathophysiology across clinical disease states in adults with DS.
For more information, contact Natalie Edwards in the Brickman Lab.

Julie McInvale
MD/PhD Student
Induced Pluripotent Stem Cell-Derived Neurons from Patients with Frontotemporal Dementia Demonstrate Altered Oxygen Consumption and Increased Susceptibility to Mitochondrial Stress
Julie J McInvale1,2,4, Sam Kwon1,2, Matti Lam1,3, Osama Al-Dalahmah2, Andrew. A. Sproul1,2, Markus D. Siegelin2, Philip L. De Jager1,3, James E. Goldman2, Vilas Menon1,3, Peter Canoll2, and Gunnar Hargus1,2
1 Taub Institute for Research on Alzheimer’s Disease and the Aging Brain, Vagelos College of Physicians and Surgeons, Columbia University Irving Medical Center, New York, NY, USA
2 Department of Pathology and Cell Biology, Vagelos College of Physicians and Surgeons, Columbia University Irving Medical Center, New York, NY, USA
3 Center for Translational & Computational Neuroimmunology, Department of Neurology, Columbia University Irving Medical Center, New York, NY, USA
4 Medical Scientist Training Program, Vagelos College of Physicians and Surgeons, Columbia University Irving Medical Center, New York, NY, USA
Frontotemporal dementia (FTD) is a heterogeneous group of incurable disorders which represent the second most common early-onset dementia, after Alzheimer's disease. In FTD patients carrying a mutation in the microtubule-associated protein tau (MAPT), the neuropathological characteristics include deposition of hyperphosphorylated tau in various brain regions, though the exact mechanisms leading to extensive neurodegeneration are unknown. Neuroinflammation and metabolic stress are emerging as common mediators of neurodegeneration across diseases.
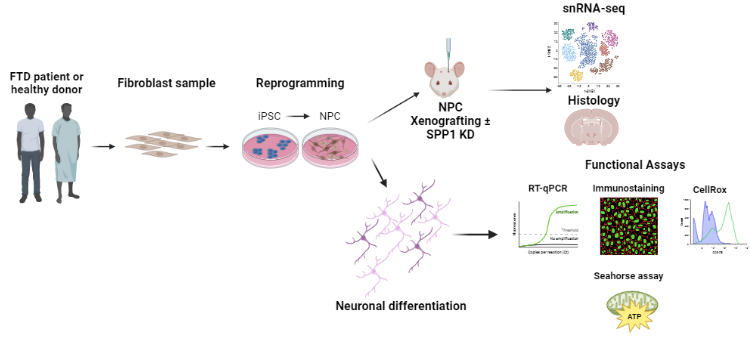
Figure 1. Schematic of research strategy. Fibroblasts from patients or healthy controls were reprogrammed to inducible pluripotent stem cells (iPSCs), with or without CRISPR-Cas9-mediated correction of the MAPT N279K mutation. iPSCs were subsequently differentiated into neural progenitor cells (NPCs) and then neurons for in vitro experiments. Control NPCs with or without knockdown of secreted phosphoprotein 1 (SPP1) were also xenografted into the forebrains of immuno-deficient mice and allowed to differentiate in vivo, followed by characterization.
Here we report via single-nucleus RNA sequencing that post-mortem neurons from FTD patients carrying the MAPT-N279K mutation show increased expression of known pro-inflammatory molecules including secreted phosphoprotein 1 (SPP1), which is recapitulated in induced pluripotent stem cell (iPSC)-derived FTD neurons harboring the same MAPT-N279K mutation. FTD neurons also showed an enhanced basal oxygen consumption rate, increased maximal respiration, and increased production of reactive oxygen species (ROS). ROS production and expression of SPP1 were attenuated after exposure to the antioxidant and ROS-blocking compound N-acetyl cysteine. FTD neurons also showed increased vulnerability to cell death after application of mitochondrial toxins. Knockdown of SPP1 in FTD neurons resulted in decreased vulnerability to oligomycin, an electron transport chain complex V toxin. Xenotransplantation of neural progenitor cells into the forebrains of mice showed that knockdown of SPP1 enhanced transplanted cell survival and integration into the neuropil and reduced reactive microgliosis at the graft-host interface. Overall, these results establish a novel link between SPP1, metabolic stress, and cell survival in the context of FTD. Future work will continue to characterize the effect of SPP1 on measures of neurodegeneration and neuronal function.
For more information, please contact Dr. Gunnar Hargus.
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.