
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspective:
August 2022
1: Retromer Deficiency in Tauopathy Models Enhances the Truncation and Toxicity of Tau
2: Aβ42 Oligomers Trigger Synaptic Loss Through CAMKK2-AMPK-Dependent Effectors Coordinating Mitochondrial Fission and Mitophagy
2: Aβ42 Oligomers Trigger Synaptic Loss Through CAMKK2-AMPK-Dependent Effectors Coordinating Mitochondrial Fission and Mitophagy
Retromer Deficiency in Tauopathy Models Enhances the Truncation and Toxicity of Tau
 |
 |  | |
| Scott Small, MD | Ismael Santa-Maria Perez, PhD | Brian D. McCabe, PhD |
Alteration of the levels, localization or post-translational processing of the microtubule-associated protein Tau is associated with many neurodegenerative disorders. The retromer complex is an evolutionarily conserved protein assembly that plays an essential role in the retrograde transport of proteins and associated lipids from endosomes to the trans-Golgi network and in the recycling of this cargo from endosomes back to the cell surface. Consistent with an association of retromer with Alzheimer’s disease, retromer activity is reduced in the entorhinal cortex of late onset AD patients. However, the molecular mechanisms through which retromer depletion may contribute to AD are poorly understood.
In collaboration with Dr. Brian McCabe’s team in the EPFL Brain Mind Institute, together with Taub colleague Dr. Scott Small, we recently published a study in Nature Communications that shows how malfunctioning of the retromer complex can lead to the buildup of toxic forms of Tau and neurodegeneration. Using both in vivo Drosophila models and cell models researchers, we found that reduction of retromer activity induces a potent enhancement of Tau toxicity through a specific and singular increase in the production of a C-terminal truncated isoform of Tau.
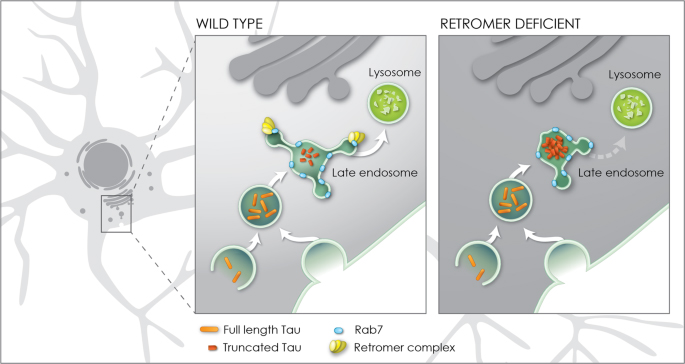
Figure 8. Model for enhancement of Tau toxicity and truncation by retromer depletion. In wildtype neurons full length Tau (orange) enters the endosomal network and is efficiently trafficked through the late endosome to the lysosome enabled by the interaction of the retromer complex (yellow) with the late endosome protein Rab7 (blue). When retromer or Rab7 activity is reduced, endosomal-lysosomal flux is retarded, exposing Tau to endosomal resident caspase activity which results in Tau truncation (red) and accumulation in the late endosome. Truncation of Tau enhances its neurotoxicity.
Our investigations further demonstrated that the interaction of the retromer complex with the late endosome protein Rab7 is critical for the enhancement of Tau-dependent neurodegeneration. Together, these results establish a molecular and cellular rationale causally linking deficient retromer activity with enhanced Tau toxicity. Deficient retromer activity slows down the trafficking of Tau allowing caspases to cut tau into a shorter form that can damage neurons. Thus, identifying drugs that enhance the trafficking of Tau might help reduce neurotoxicity and tauopathy.
Ismael Santa-Maria Perez, PhD
Assistant Professor of Pathology and Cell Biology (in the Taub Institute)
is2395@cumc.columbia.edu
 |
 |  | |
| Andrew A. Sproul, PhD | Franck Polleux, PhD | Karen Duff, PhD |
Dysfunction of mitochondria, organelles responsible for energy production in mammalian cells including neurons, has been implicated in Alzheimer’s disease (AD) pathogenesis by a growing body of literature. In a recent publication in Nature Communications by senior author and Columbia University researcher Dr. Franck Polleux, it was shown that synaptic spine loss caused by soluble Aβ oligomers in the AD J20 mouse model (APP Swedish and Indiana mutant overexpression) is mediated by mitochondrial remodeling driven by overactivation of AMPK, and requires phosphorylation of tau on ser262. Elevated AMPK signaling promotes mitochondrial fission via phosphorylation of MFF (mitochondrial fission factor) and mitophagy via phosphorylation of ULK2 (autophagy activating kinase 2), with a net result of the loss of mitochondrial mass in neurons affected the earliest in the AD brain. Inhibition of this pathway, by a variety of means, protected against spine loss.
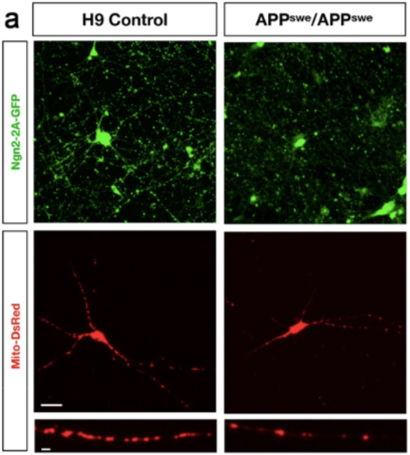
Figure: Analysis of mitochondria in APP Swedish and isogenic control human neurons.
In order to extend some of these findings into a human-specific experimental system, my laboratory collaborated with Dr. Polleux and his group, utilizing our novel APP Swedish (KM670/771NL) human embryonic stem cell (hESC) model. This model was generated by using CRISPR/Cas9 genome-editing technology to introduce the APP Swedish mutation into both alleles of the control H9 line, the most commonly used hESC line including use in cell replacement-focused clinical trials. To our knowledge, this is the first AD hESC model published. APP Swedish neurons had reduced mitochondrial biomass and elevated AMPK signaling relative to isogenic control neurons, consistent with findings in AD mutant mice.
The excitatory human neurons produced for this study were generated using a novel permanent-line method developed by our group and published last year in Current Protocols. Human pluripotent stem cell lines are either infected with lentiviral particles encoding Ngn2 (and co-doxycycline activator rtTA) to make excitatory neurons, or ASCL1 and DLX2 (along with rtTA) to make inhibitory neurons. Antibiotic-selected permanent lines are propagated as stem cells and terminally differentiated to neurons by addition of doxycycline and neural media. These methods were developed by my lab in an active collaboration with Dr. Karen Duff, to help investigate why excitatory neurons are more vulnerable to tauopathy than inhibitory neurons.
Andrew A. Sproul, PhD
Assistant Professor of Pathology and Cell Biology (in the Taub Institute) at CUMC
aas2003@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.