
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspective:
April 2024
1: Rare Genetic Variation in Fibronectin 1 (FN1) Protects Against APOEε4 in Alzheimer's Disease
2: Cell Subtype-Specific Effects of Genetic Variation in the Alzheimer's Disease Brain
3: Osteopontin Drives Neuroinflammation and Cell Loss in MAPT-N279K Frontotemporal Dementia Patient Neurons
4: Childhood Engagement in Cognitively Stimulating Activities Moderates Relationships Between Brain Structure and Cognitive Function in Adulthood
2: Cell Subtype-Specific Effects of Genetic Variation in the Alzheimer's Disease Brain
3: Osteopontin Drives Neuroinflammation and Cell Loss in MAPT-N279K Frontotemporal Dementia Patient Neurons
4: Childhood Engagement in Cognitively Stimulating Activities Moderates Relationships Between Brain Structure and Cognitive Function in Adulthood
Rare Genetic Variation in Fibronectin 1 (FN1) Protects Against APOEε4 in Alzheimer's Disease
 |
 |  | |
| Prabesh Bhattarai, PhD | Tamil Iniyan Gunasekaran, PhD | Michael E. Belloy, PhD | |
 |
 |  | |
| Richard Mayeux, MD, MSc | Caghan Kizil, PhD | Badri N. Vardarajan, PhD, MS |
Presence of the APOEε4 allele is the biggest confirmed genetic risk factor for late-onset Alzheimer’s disease (AD). Homozygosity for the APOEε4 is associated with increased risk and nearly complete penetrance. However, some individuals carrying APOEε4 can defy the onset of dementia, and the mechanisms of this protection remain largely unknown. Our collaborative team from the Taub Institute hypothesized that elderly APOEε4 carriers that “escaped” the onset of AD might harbor genetic variants that protect them or delay the onset of disease. To interrogate this hypothesis, we analyzed whole genome sequencing data in 3,500 individuals from over 700 multiplex families in the EFIGA and NIA-FBS cohorts, to identify genetic variants that protect or delay the onset of disease.
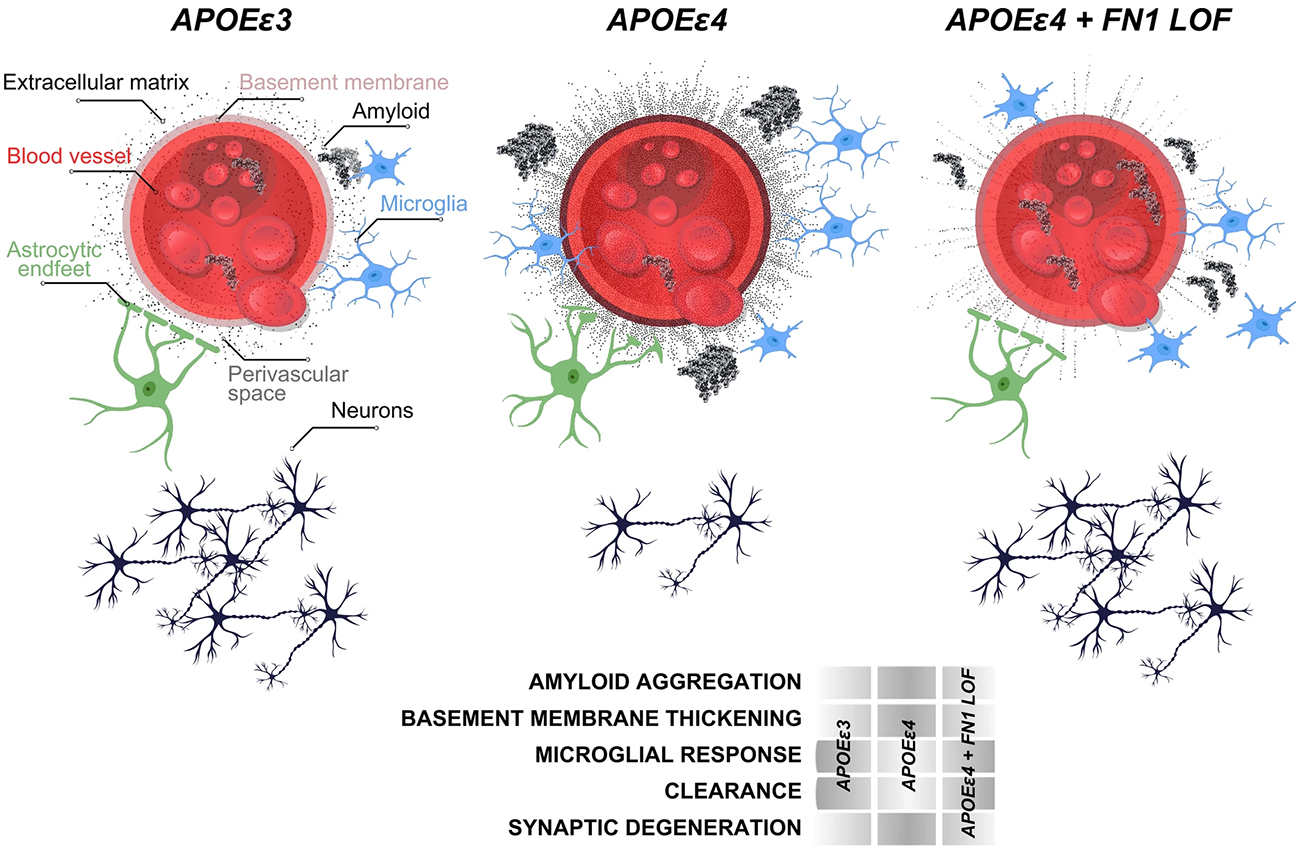
Figure. Schematic abstract for the protective effect of FN1 variants.
As recently published in Acta Neuropathologica and featured in the CUIMC Newsroom, our study identified 510 genetic variants that might provide resilience. The genes containing these variations significantly enriched the biological pathways that were related to the extracellular matrix around the blood-brain-barrier (BBB). Our team prioritized a genetic variant in the gene for fibronectin (FN1). This finding was substantiated by our Columbia team and independently corroborated by colleagues from Stanford and Washington universities, solidifying the findings across a significant sample size of 11,000 participants. The genetic variant in FN1 diminished the pathological accumulation of Fibronectin, and gliosis around the blood vessels in humans. This FN1 variant reduced the odds of developing Alzheimer’s by 71% and delayed disease onset by four years.
Functional studies in FN1 knockout zebrafish models, alongside analysis of postmortem human brains, helped to identify the mechanisms by which the loss of function in fibronectin safeguards against AD. Essentially, FN1 loss-of-function enhanced the clearance of amyloid and microglial activity. Therapeutically, the fibronectin variant is an enticing target, likely to inspire new drug development avenues. By mimicking the variant’s protective effect, future treatments could design defense against AD. This discovery also provides significant impulse for the therapeutic focus toward vascular components of the brain, and highlights the BBB's pivotal role in AD progression.
Badri N. Vardarajan, PhD, MS
Assistant Professor of Neurological Science (in Neurology, the Gertrude H. Sergievsky Center, and the Taub Institute)
bnv2103@cumc.columbia.edu
Caghan Kizil, PhD
Associate Professor of Neurological Sciences (in Neurology and the Taub Institute)
ck2893@cumc.columbia.edu
Richard Mayeux, MD, MSc
Gertrude H. Sergievsky Professor of Neurology, Psychiatry and Epidemiology (in the Taub Institute and the Gertrude H. Sergievsky Center)
rpm2@cumc.columbia.edu
Cell Subtype-Specific Effects of Genetic Variation in the Alzheimer's Disease Brain
 |  |  | ||
Masashi Fujita, PhD | Vilas Menon, PhD | Philip L. De Jager, MD, PhD |
Genome-wide association studies (GWAS) have successfully identified genetic risk variants associated with disease susceptibility in neurodegenerative and neuropsychiatric conditions, including Alzheimer's disease (AD). Previous investigations have revealed that many of these risk variants affect the expression level of nearby genes in samples of homogenized cortical tissue in which all cell types are mixed together. However, the human neocortex contains an intricate cellular landscape that is composed of diverse neuronal and glial cell types, each with distinct subtypes. Thus, our perspective on the functional consequences of Alzheimer disease susceptibility variants was limited. To date, expression Quantitative Trait Locus (eQTL) analyses, in which one maps the effect of genetic variants on the expression of nearby genes, had not interrogated the effect of genetic variation in this context and had failed to capture the cellular specificity of gene expression in the different cell types and subtypes of the brain.
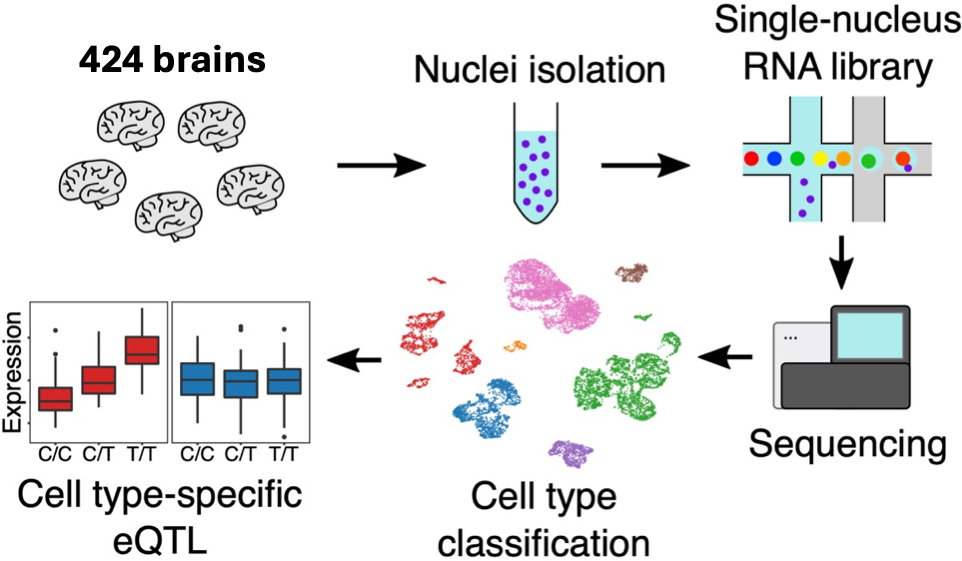
Figure 1. Schema of our study.
To address this critical gap, our team sequenced over 1.6 million individual cells from samples of the frontal cortex of 424 human brains, and we integrated these data with whole genome sequence data available from each individual. These data were then analyzed to produce a detailed map of eQTL effects in each of the 6 main neuronal and glial cell type as well as their 92 cell subtypes. The results were recently published in Nature Genetics. Specifically, we identified 10,004 eGenes (genes which are the target of the eQTL effect) at the cell type level and 8,099 eGenes at the cell subtype level. Many eGenes were exclusively detected within specific cell subtypes, particularly in neurons. This underscores the critical importance of subtype-level analyses in future single-cell eQTL studies. An intriguing example of eQTL discovery was a microglia-specific eQTL associated with the APOE gene, the gene with the strongest genetic risk factor for AD. Despite being located 40 kb away from the APOE gene body, this variant significantly influenced APOE expression only in microglia, the resident immune cell of the brain. Remarkably, this SNP is associated with AD risk according to recent GWAS studies and correlates with the level of cerebral amyloid angiopathy. This intersection of genetics, microglia biology, and disease pathology presents a promising avenue for future investigation.
To leverage our eQTL resources to gain insights into disease mechanism, we compared our eQTL with publicly available GWAS summary statistics of AD and other neurodegenerative diseases. Colocalization analyses of eQTL and GWAS identified a large number of AD susceptibility variants that also influence gene expression in specific brain cell types, revealing the functional consequence, at the molecular level, of those AD variants. It also prioritizes the target cell type and subtype of interest for each AD variant. Interestingly, these analyses reveal that microglia have an excess of these functional consequences relative to other cell types, highlighting their pre-eminent role in initiating the cascade of pathologic events that ultimately lead to AD. However, our resource is not disease-specific, and we performed similar analyses for other neurodegenerative conditions, including amyotrophic lateral sclerosis, Parkinson's disease, and schizophrenia as well as other brain-related such educational attainment. All data and results have been shared through the AD Knowledge Portal which is supported by the national Institute of Aging. Thus, our work not only provided important new insights into the earliest molecular events that ultimately lead to AD and other neurologic diseases but also facilitates repurposing of our data in collaborative efforts that will continue to accelerate discoveries in neurology. The implication of microglial APOE expression in cerebral amyloid angiopathy, an AD-related pathology that contributes to dementia, is particularly salient and may influence the risk of some of the new anti-amyloid treatments for AD; this promises to be an important observation that will drive further investigation.
Philip L. De Jager, MD, PhD
Weil-Granat Professor of Neurology (in Neurology and the Taub Institute)
pld2115@cumc.columbia.edu
 |  | |
| Osama Al Dalahmah, MD, PhD | Matti Lam, PhD | |

|  | |
| Julie McInvale, MD/PhD Student | Gunnar Hargus, MD, PhD |
Frontotemporal dementia (FTD) is a group of neurodegenerative diseases and a major cause of dementia in patients under 60 years of age without any treatment options. About half of patients have widespread deposition of hyperphosphorylated tau (p-tau) protein in various brain regions including frontal and temporal lobes as well as basal ganglia and brain stem (FTD-tau). A family history is seen in about half of the patients with FTD, and 10-30% of these patients carry autosomal-dominant mutations in MAPT on chromosome 17q21 encoding tau. There is evidence that neuroinflammation plays an important role in the pathogenesis of FTD. As such, microglial activation is found in areas of neuronal loss and increased p-tau burden, and in close proximity to p-tau containing cells in patients and animal models of FTD.
In our most recent study published in Cell Stem Cell, we made the interesting discovery that FTD neurons contribute to microglial activation via the release of the proinflammatory protein osteopontin (OPN). For this project, we performed single nucleus RNA-sequencing (snRNA-seq) on postmortem brain tissue from FTD patients carrying the MAPTN279K mutation and from control individuals revealing a strong pro-inflammatory gene expression signature and disturbed mitochondrial function in FTD neurons (Figure 1; box 1). We had generated induced pluripotent stem cells (iPSCs) from FTD patients carrying the same MAPTN279K mutation and identified disease phenotypes in FTD neurons related to MAPT splicing, tau phosphorylation, oxidative stress, production of reactive oxygen species (ROS), mitophagy, oxidative phosphorylation and neuroinflammation with an upregulation and release of OPN into the medium supernatant (Figure 1; boxes 2 and 3). OPN was also highly upregulated in postmortem FTD neurons and it activated cultured human microglia that we had differentiated from both FTD and control iPSCs.
.jpg)
Figure 1. Graphical summary of this study.
When we transplanted FTD and control NPCs into the forebrain of immunocompromised mice, we found that FTD neurons survived less, formed smaller grafts and showed impaired outgrowth into the surrounding brain parenchyma (Figure 1; boxes 4 and 5). In addition, the FTD neurons elicited an increased microglial response four months after injection of cells. We performed snRNA-seq analyses of microdissected FTD and control grafts, which captured both human and mouse gene expression signatures. These analyses validated our observations of neuronal pathology and cell stress in FTD neurons, and they also validated our findings of increased reactivity in mouse microglia. When we overexpressed OPN in transplanted iPSC-derived neurons, there was a strong microgliosis at the injection site and the human cells did not survive. Conversely, downregulation of OPN in engrafted FTD neurons resulted in improved engraftment and in reduced microglial infiltration (Figure 1; box 5). These findings indicated an immune-modulatory role of OPN in FTD patient neurons, which may represent a potential therapeutic target in FTD.
This study was performed in collaboration with Drs. Vilas Menon, Phil De Jager, James Goldman and Andrew Sproul from the Taub Institute and with Drs. Peter Canoll, Markus Siegelin and Claudia Doege from the Department of Pathology and Cell Biology at CUIMC.
Gunnar Hargus, MD, PhD
Assistant Professor of Pathology and Cell Biology
gh2374@cumc.columbia.edu
 |  | |
Alexandra Gaynor, PhD | Yian Gu, MD, MS, PhD |
Previous research suggests greater engagement in cognitively stimulating activities (CSA) may protect against neurocognitive decline. However, previous studies have focused on CSA during childhood, while no research to date has investigated whether CSA during childhood and changes protects against the effects of brain changes on cognition later in life.
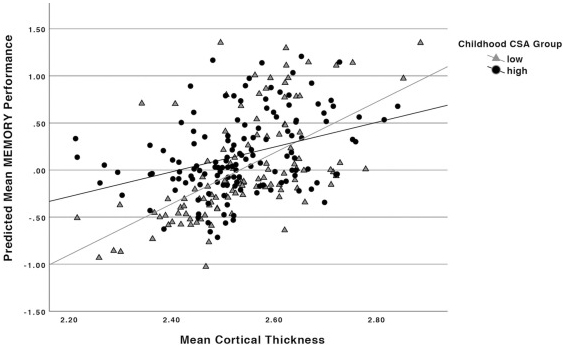
Figure 1. Relationship between baseline mean cortical thickness and memory performance by childhood CSA group. Results of multivariable linear regressions modeling interaction between childhood CSA group and baseline mean cortical thickness on baseline memory performance. Models adjusted for effects of age, education, gender, race, and IQ.
Together with former postdoctoral fellow and lead author Dr. Alexandra Gaynor (now at Montclair State University) and colleagues from Taub, we tested the moderating role of childhood CSA in relationships between brain structure and cognitive performance during adulthood. A total of 250 healthy adults aged 20-80 years underwent MRI to assess brain structural measures and completed neuropsychological tests to measure cognitive functions on perceptual speed, fluid reasoning, and episodic memory, at two time points 5 years apart. Participants also self-reported a validated questionnaire inquiring about childhood CSA, including nine cognitive activities during childhood. A composite mean childhood CSA score was calculated and then categorized into low and high levels of engagement in CSA during childhood.
As published last month in Neurobiology of Aging, we found that the impact of 5-year brain change (in cortical thickness and brain volume) on cognitive decline (in processing speed) was smaller in individuals with high childhood CSA than those with low childhood CSA. These findings suggest higher CSA during childhood may help maintain cognitive abilities in face of brain structure changes later in life. Further research is needed to understand how factors that may be associated with childhood CSA, such as socioeconomic status, impact neurocognitive aging later in life. Nonetheless, these findings offer important insight into the potential protective factors early in life that may attenuate the negative sequelae of decreasing structural integrity during adulthood.
Yian Gu, MD, MS, PhD
Associate Professor of Neurological Sciences (in Neurology, Epidemiology, the Gertrude H. Sergievsky Center, and the Taub Institute)
yg2121@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.