
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspective:
January 2023
1: Histopathology of the Cerebellar Cortex in Essential Rremor and Other Neurodegenerative Motor Disorders: Comparative Analysis of 320 Brains
2: The Caribbean-Hispanic Alzheimer's Disease Brain Transcriptome Reveals Ancestry-Specific Disease Mechanisms
3: Comparison of Amyloid Burden in Individuals with Down Syndrome Versus Autosomal Dominant Alzheimer's Disease: A Cross-Sectional Study
4: Neuronal Membrane Proteasomes Regulate Neuronal Circuit Activity in Vivo and are Required for Learning-Induced Behavioral Plasticity
2: The Caribbean-Hispanic Alzheimer's Disease Brain Transcriptome Reveals Ancestry-Specific Disease Mechanisms
3: Comparison of Amyloid Burden in Individuals with Down Syndrome Versus Autosomal Dominant Alzheimer's Disease: A Cross-Sectional Study
4: Neuronal Membrane Proteasomes Regulate Neuronal Circuit Activity in Vivo and are Required for Learning-Induced Behavioral Plasticity
 |
 |
 | ||
| Sheng-Han Kuo, MD | Jean Paul G. Vonsattel, MD | Phyllis L. Faust, MD, PhD |
Until recently, the pathological anatomy of essential tremor (ET) had been largely unexplored, a fact that is surprising given the high prevalence of this disease. In recent years, we have identified a growing compendium of morphologic changes in the ET brain, predominantly centered in and around the Purkinje cells (PCs) in the cerebellar cortex, and distinguishing ET from age-matched control brains. These changes affected the PC dendritic arbor, the PC axonal compartment, the PC soma as well as the connections between PCs and neighboring neuronal populations (climbing fibers, basket cells). However, the degree to which these same changes occur in other diseases, either those that are cerebellar degenerative [e.g., spinocerebellar ataxias (SCA), Friedreich’s ataxia (FA), multiple system atrophy (MSA)] or those that are more broadly degenerative but physiologically also involving the cerebellum and manifesting tremor [e.g., Parkinson’s disease (PD), dystonia], or are unique to ET was not clear.
In a study published in Acta Neuropathologica, in collaboration with Dr. Elan Louis (UT Southwestern), Dr. Sheng-Han Kuo in Neurology and Taub colleague Dr. Jean Paul Vonsattel, we designed a novel approach using statistical comparisons based on composite analyses of multiple morphological metrics in a postmortem series of 320 brains on the severity and patterning of cerebellar cortex changes in ET (n = 100), SCAs (n = 47, including 13 SCA3 and 34 SCA1, 2, 6, 7, 8, 14); FA (n = 13); MSA (n = 29); PD (n = 62); and dystonia (n = 19) versus controls (n = 50). Using quantitative data on 37 pathological metrics, only a few changes were observed in dystonia and PD, whereas the pathological changes were generally on the milder end of a degenerative spectrum in ET, FA and SCA3, and on the more severe end of that spectrum in SCA1/2/6/7/8/14 and MSA. In large part, the changes observed in ET and the other cerebellar degenerative disorders comprised differences of degree rather than kind, likely reflecting a stereotypic repertoire of cellular reactions that characterize cerebellar degeneration. In addition, features distinctive to specific disorders were also identified, including a redistribution of climbing fiber synapses to the outer PC dendritic arbor in ET and FA, and in ET this was uniquely combined with CF synaptic regression.
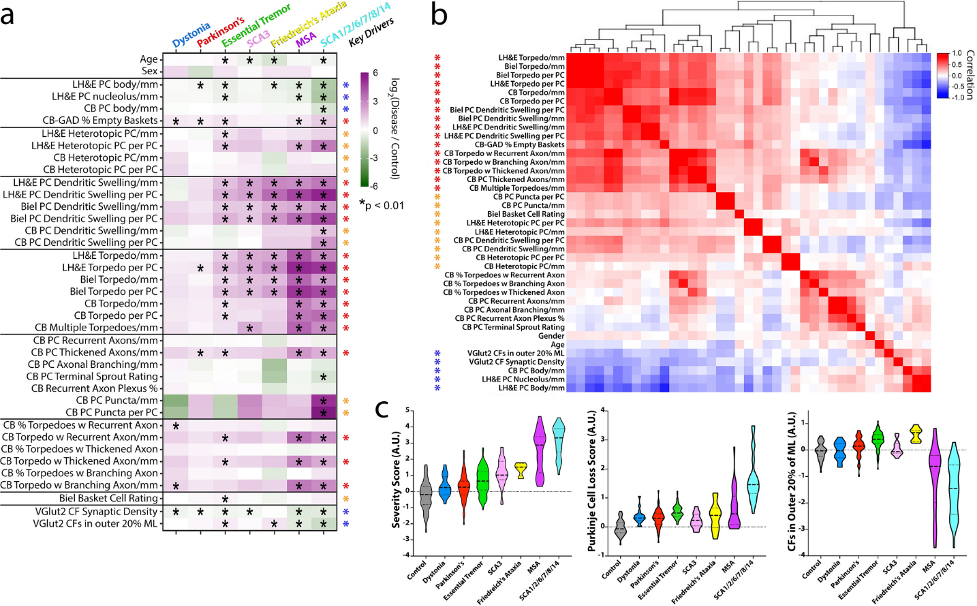 Figure 2. Differences in fold-change for each metric across diseases and key pathologic drivers. a Metrics (rows) and diagnoses (columns) are shown. Each cell in the heatmap is the average log2-fold-change (disease vs. control) for each metric. The scale ranges from dark purple (high relative to control) to dark green (low relative to control). Elements with a black asterisk indicate a statistically significant difference vs. controls (Mann–Whitney test, p values corrected for false discovery by Benjamini–Hochberg method; * = FDR< 0.01). b Hierarchical clustering of the correlation coefficients between all pairwise combinations of metrics, with colored scale of red (positive correlation), white (no correlation) and blue (negative correlation). Many of the prominent positively correlated (red and orange asterisks) or negatively correlated (blue asterisk) variables are the same variables that differ across disease categories (see black asterisks in 2a), and are designated here as “Key Drivers” in (a). c Three scores were computed across disease categories by combining the average fold-change (disease/control) for selected metrics and plotted on a log2-transformed scale, including a “Severity Score” (large block of 25 positively correlated red and orange metrics in panel (b)), a “Purkinje Cell Loss Score” (inverse Purkinje cell body and percent empty baskets), and a score reflecting climbing fibers (CFs) in the outer 20% of the molecular layer. Within each violin, the dashed line shows the median value and the dotted lines indicate outer quartiles in the data distribution. Data for Friedreich’s ataxia was derived from 4 cases in whom metrics from calbindinD28k staining was performed and 13 cases for all other metrics performed in paraffin sections.
|
Our study expands the knowledge of cerebellar pathological changes across neurodegenerative motor disorders and provides a robust method to identify disease patterns in human postmortem tissue. There is some evidence that all these motor disorders do not blandly express the same generic pattern of degeneration, with their only distinguishing feature being the degree to which they express that pattern, and that a somewhat distinctive signature of degenerative changes marks each of these disorders.
Phyllis L. Faust, MD, PhD
Professor of Pathology and Cell Biology at the CUMC
plf3@cumc.columbia.edu
 |  | |
| Daniel Felsky, PhD | Ismael Santa-Maria Perez, PhD | |

|  | |
| Caghan Kizil, PhD | Giuseppe Tosto, MD, PhD |
Diversity in postmortem brain studies of Alzheimer’s Disease (AD) is lacking. Identifying ancestry-specific molecular profiles is indeed crucial to understand novel mechanisms and develop effective interventions in non-European, high-risk populations. In this study, we performed gene differential expression and consensus network-based analyses in RNA-sequencing data of postmortem brain tissue (temporal cortex) from 39 Caribbean Hispanics derived from the Columbia University Brain Bank. We also used two large non-Hispanic White cohorts (the Religious Orders Study and Memory and Aging Project and the Mayo Clinic RNAseq cohort) to compare results across different ancestries.
As recently reported in Neurobiology of Disease, we identified 2,802 significantly differentially expressed genes in Caribbean Hispanics, including several AD known-genes. Overall, the differential expression results were highly concordant across ethnicities, with 373 genes transcriptome-wide significant in all three cohorts. Transethnic expression meta-analysis found NPNT to be the top differentially expressed gene. We also aimed to identify genes with ancestry-specific effects: 118 genes showed significant effects in Caribbean Hispanics and an opposite direction of effect in both non-Hispanic White cohorts. Among the strongest ancestry-specific genes was CSPG4, a chondroitin sulfate proteoglycan that is expressed in oligodendrocyte precursor cells and a subpopulation of NG2+ astrocytes, which has been strongly linked to AD pathology in humans and mice.
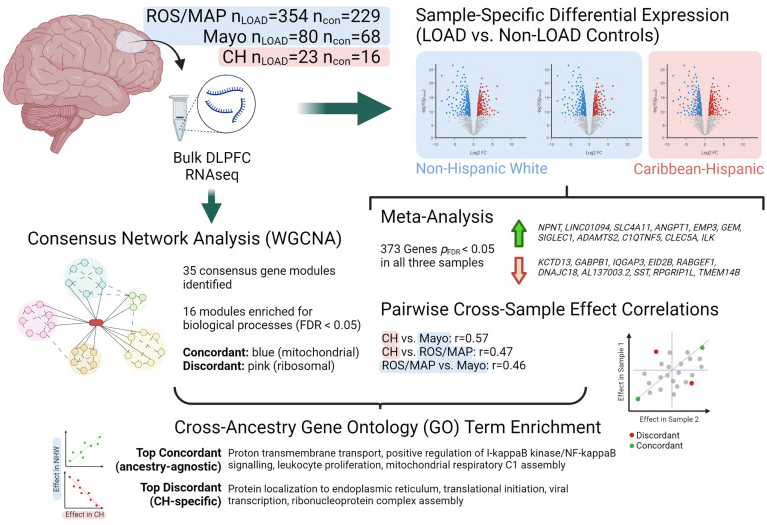 Figure 1.Schematic showing study design and top results.
CH = Caribbean Hispanic; con = non-LOAD control; DLPFC = dorsolateral prefrontal cortex; FDR = false discovery rate; LOAD = late-onset Alzheimer's disease; logFC = log fold-change; ROS/MAP = Religious Orders Study / Memory and Aging Project; WGCNA = weighted gene correlation network analysis. Created with BioRender.com.
|
We also analyzed high-depth single-nucleus RNAseq data from entorhinal cortex and superior frontal gyrus to localize our findings to specific brain cell types. Out of the 1,050 genes that were gene-wide significant in the transethnic metanalysis, 829 genes were differentially expressed in at least one cell type in the single cell data. These genes included the top hit NPNT. Interestingly, 69 out of 75 known AD-known genes were also differentially expressed in at least one cell type, including SORL1, ABCA7, PLCG2, CLU, FBXL7, UNC5C. For instance, ABCA7 was found to be differentially expressed in neurons only, while FBXL7 was differently expressed in all cell types but microglia. Importantly, FBXL7 was first reported by our group as the top signal in the largest Caribbean Hispanic GWAS performed to date.
Increasing representation in genetic studies will allow for deeper understanding of ancestry-specific mechanisms and improving precision treatment options in understudied groups.
Giuseppe Tosto, MD, PhD
Assistant Professor of Neurological Sciences (in Neurology, the Taub Institute and the Gertrude H. Sergievsky Center)
gt2260@cumc.columbia.edu
 |
 |
| Joseph H. Lee, DrPH | Adam M. Brickman, PhD |
Individuals with Down syndrome (DS) have an extra copy of the APP gene and overproduce amyloid ß (Aß), placing them at especially high risk for developing Alzheimer’s disease (AD). It remains unclear, however, whether the timing and spatial distribution of amyloid accumulation differs between people with DS versus individuals with other forms of AD, including those with autosomal dominant AD. Therefore, as part of the latest effort from the multi-site Alzheimer's Biomarker Consortium-Down Syndrome (ABC-DS) study, our team of investigators, including Taub colleague Dr. Joseph Lee, evaluated amyloid deposition in these two groups with genetic causes of Alzheimer's disease (Down syndrome vs autosomal dominant).
As recently reported in The Lancet Neurology, 192 individuals with DS and 33 sibling controls from the ABC-DS study and 265 carriers of autosomal dominant AD mutations and 169 non-carrier familial controls from the DIAN study were included in our analyses. This is the first known study to assess amyloid PET in relation to CSF amyloid concentrations in people with DS, to understand the relationship between amyloid clearance and deposition. We also assessed global and regional amyloid PET as a function of cognitive performance and age for individuals with DS and autosomal dominant AD.
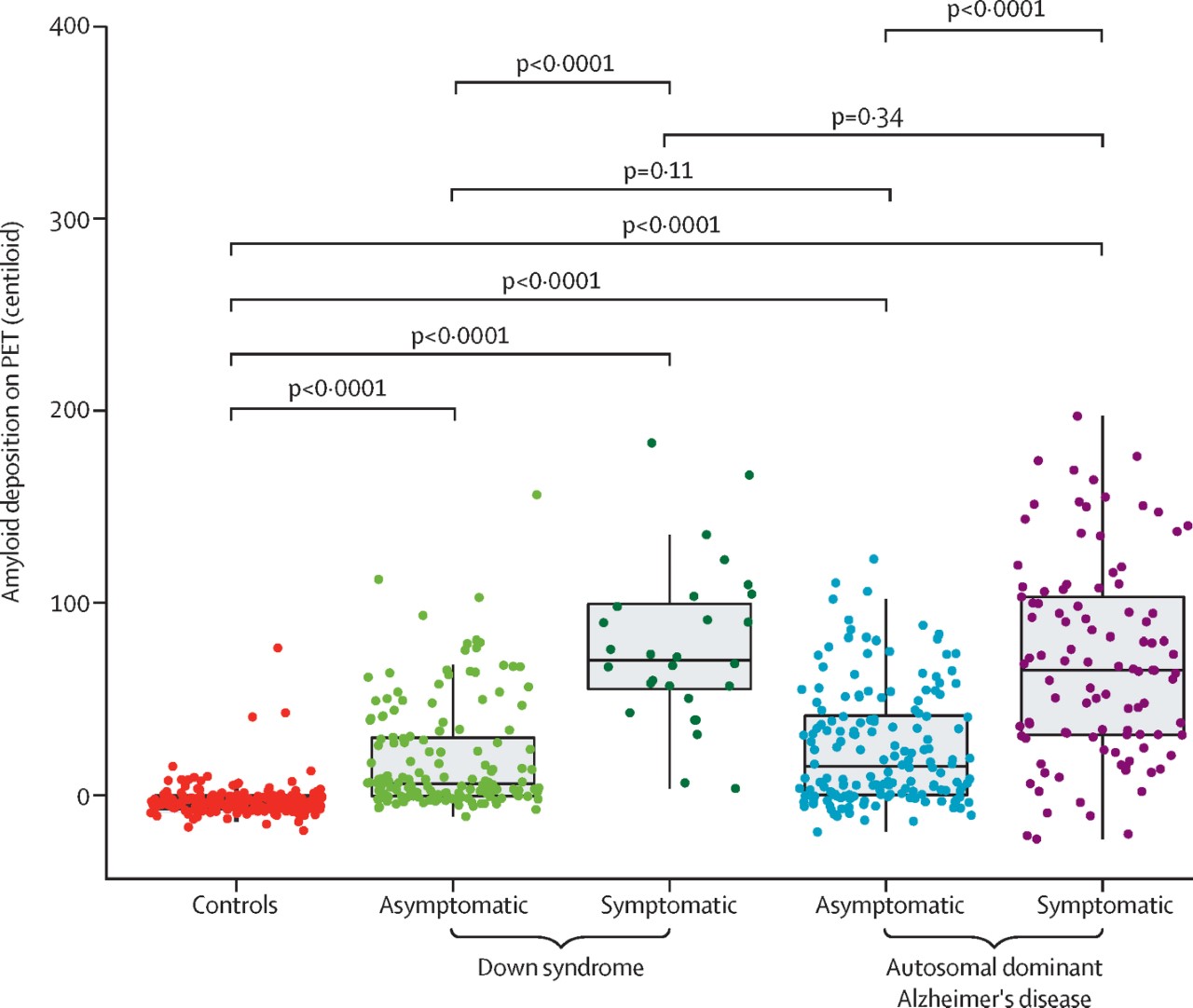
Figure 2. Global amyloid deposition on PET by cognitive status. Controls, n=202; asymptomatic Down syndrome, n=155; symptomatic Down syndrome, n=28; asymptomatic autosomal dominant Alzheimer's disease, n=164; symptomatic autosomal dominant Alzheimer's disease, n=101.
Overall, our group observed many similarities between these groups. Our findings showed an inverse relation between these two amyloid measures, suggesting that amyloid concentrations decrease in the CSF with accumulating amyloid in the brain in people with DS, similar to the pattern seen in people with late-onset AD and autosomal dominant AD. Although our findings suggest that amyloid begins accumulating at the early stages of AD progression in both groups, we also observed that amyloid accumulation began earlier in carriers of autosomal dominant AD mutations than in people with DS but approached similar concentrations at the estimated symptom onset. After differentiating mutation carriers by autosomal dominant mutation type, we found that this difference in timing was mainly driven by PSEN1 mutations.
In conclusion, this study shows that, although there are subtle differences, Alzheimer's disease pathophysiology is similar among people with Down syndrome and people with autosomal dominant Alzheimer's disease. These subtle regional differences in amyloid deposition between people with DS, autosomal dominant AD, and late-onset AD are important to consider for future clinical trials. Different brain regions might need to be evaluated when determining amyloid-positivity and the efficacy of anti-amyloid therapies for these groups.
Adam M. Brickman, PhD
Professor of Neuropsychology (in Neurology, the Taub Institute and the Gertrude H. Sergievsky Center)
amb2139@cumc.columbia.edu

Kapil V. Ramachandran, PhD
All cells have developed extensive and elaborate mechanisms to regulate protein homeostasis. The collective actions of the protein homeostasis machinery determine cellular capacity to defend against protein aggregates, which are frequently and commonly observed in a wide variety of neurodegenerative disorders. One of the fundamental mechanisms which maintain protein homeostasis in all cells is the ubiquitin-proteasome system (UPS). Prior to joining Columbia, our group discovered a new mechanism of protein degradation in the nervous system. This relies on a proteasome which is distinct from the classic UPS, where the core part of the proteasome is localized to the neuronal plasma membrane and by all measures, traverses the plasma membrane.
One of the fundamental characteristics of this so-called neuroproteasome is that substrates are degraded across the membrane into release extracellular polypeptides. We hypothesized that these small peptides are used as a new mechanism of cell-cell communication. In our initial work, we performed many of our experiments in primary cell cultures as a way to mechanistically interrogate this complex. Therefore, we lacked a real understanding of the function of neuroproteasomes in vivo.
When we began to think about how to attack this problem, we lacked a reasonable system to interrogate the function of neuroproteasomes in vivo. The inhibitors were prohibitively expensive to do this at scale in the mouse system and we did not even have a clear understanding of what to measure. At a meeting a few years ago, Holly Cline at Scripps Research Institute presented data in the Xenopus tadpole that inhibition of protein synthesis had very acute and rapid effects on the plasticity of tectal neurons in vivo. I thought this might be a connected mechanism to how neuroproteasomes regulate plasticity and the Xenopus system was perfect to interrogate this—small, easy to manipulate and image, clear behavioral readouts. I approached Holly at this meeting and this began a fantastic collaboration.
For the first time, Holly and her then-postdoctoral candidate and now-faculty member at Georgetown, Haiyan He, injected neuroproteasome inhibitors in vivo into Xenopus and demonstrated that neurpproteasomes regulate neuronal activity in vivo. They then extended this to find that neuronal plasticity and plasticity-dependent behaviors are occluded by neuroproteasome inhibition. As recently reported in PNAS, this cross-disciplinary and cross-species analysis not only demonstrates the conservation of neuroproteasomes, but also highlights, for the very first time, that neuroproteaosmes are indispensable for neuronal plasticity. This is important because plasticity encompasses the mechanisms underlying learning and memory, which are definitional to cognitive decline over age and in neurodegenerative disease. These data demand to be extended in mammalian systems, but also in the context of age- and neurodegeneration-related cognitive decline.
Kapil V. Ramachandran, PhD
Assistant Professor of Neurological Sciences (in Neurology, Neuroscience, and the Taub Institute)
kvr2113@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.