
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspective:
July 2022
1: GW5074 Increases Microglial Phagocytic Activities: Potential Therapeutic Direction for Alzheimer's Disease
2: Cerebral Amyloid Angiopathy Interacts with Neuritic Amyloid Plaques to Promote Tau and Cognitive Decline
3: Amyloid, Cerebrovascular Disease, and Neurodegeneration Biomarkers Are Associated with Cognitive Trajectories in a Racially and Ethnically Diverse, Community-Based Sample
2: Cerebral Amyloid Angiopathy Interacts with Neuritic Amyloid Plaques to Promote Tau and Cognitive Decline
3: Amyloid, Cerebrovascular Disease, and Neurodegeneration Biomarkers Are Associated with Cognitive Trajectories in a Racially and Ethnically Diverse, Community-Based Sample
View 2022 - 2021 Archive || 2020 Archive || 2019 Archive ||
2018 Archive || 2017 and Earlier Archive
May 2022:
April 2022:
March 2022:
February 2022:
January 2022:
December 2021:
November 2021:
October 2021:
September 2021:
August 2021:
July 2021:
June 2021:
May 2021:
April 2021:
March 2021:
February 2021:
Progranulin Mutations in Clinical and Neuropathological Alzheimer's Disease
Wolframin is a Novel Regulator of Tau Pathology and Neurodegeneration
Homotypic Fibrillization of TMEM106B Across Diverse Neurodegenerative Diseases
Correlation of Plasma and Neuroimaging Biomarkers in Alzheimer's Disease
Tubulin Tyrosination Regulates Synaptic Function and is Disrupted in Alzheimer's Disease
The Penalty of Stress - Epichaperomes Negatively Reshaping the Brain in Neurodegenerative Disorders
The Neuronal Retromer can Regulate Both Neuronal and Microglial Phenotypes of Alzheimer's Disease
Deep Learning Improves Utility of Tau PET in the Study of Alzheimer's Disease
Age of Onset of Huntington's Disease in Carriers of Reduced Penetrance Alleles
Caspase-9: A Multimodal Therapeutic Target With Diverse Cellular Expression in Human Disease
Midlife Vascular Factors and Prevalence of Mild Cognitive Impairment in Late-Life in Mexico
The Association Between Sex and Risk of Alzheimer's Disease in Adults with Down Syndrome
Marked Mild Cognitive Deficits in Humanized Mouse Model of Alzheimer's-Type Tau Pathology
Rapid ATF4 Depletion Resets Synaptic Responsiveness after cLTP
Polygenic Risk Score for Alzheimer's Disease in Caribbean Hispanics
Recognition Memory and Divergent Cognitive Profiles in Prodromal Genetic Frontotemporal Dementia
The Microtubule Cytoskeleton at the Synapse & The Synaptic Life of Microtubules
Optimizing Subjective Cognitive Decline to Detect Early Cognitive Dysfunction
The AD Tau Core Spontaneously Self-Assembles and Recruits Full-Length Tau to Filaments
Olfactory Impairment is Related to Tau Pathology and Neuroinflammation in Alzheimer's Disease
Pathogenic Role of Delta 2 Tubulin in Bortezomib-Induced Peripheral Neuropathy
 |
 |  | |
| Sarah M. Connor, MS | Mamunur Rashid, PhD | Elizabeth M. Bradshaw, PhD |
Alzheimer’s disease (AD) is a progressive disorder that destroys memory and cognition and is ultimately fatal, affecting millions of individuals with an immensely personal and social burden. It is considered among the top 10 killer diseases in the U.S. for people above 65 years with extremely limited options for drug treatments. Clinical trials using antibodies targeting the hallmark pathologies have consistently failed to rescue cognition. These antibody-based therapies utilize microglia to clear pathology, but they do not modify the microglia phenotype itself, which is thought to be dysfunctional with disease.
Human genetics indicate that a microglia-specific intervention could be a more strategic way to treat sporadic or late-onset AD (LOAD). Thus, to specifically target microglia, we performed a high content drug screen with a microglial culture model, monocyte-derived microglia-like (MDMi) cells. Previously, we showed that MDMi are genetically and phenotypically similar to human microglia making this an ideal model for drug screens with different genetic backgrounds accounted for (Ryan et al., 2017). Utilizing drug repurposing to identify potential therapeutics that target microglial phagocytosis for AD treatment, we performed a drug screen using the Library of Pharmacologically Active Compounds (LOPAC1280), a collection of 1,280 chemicals with known biological functions.
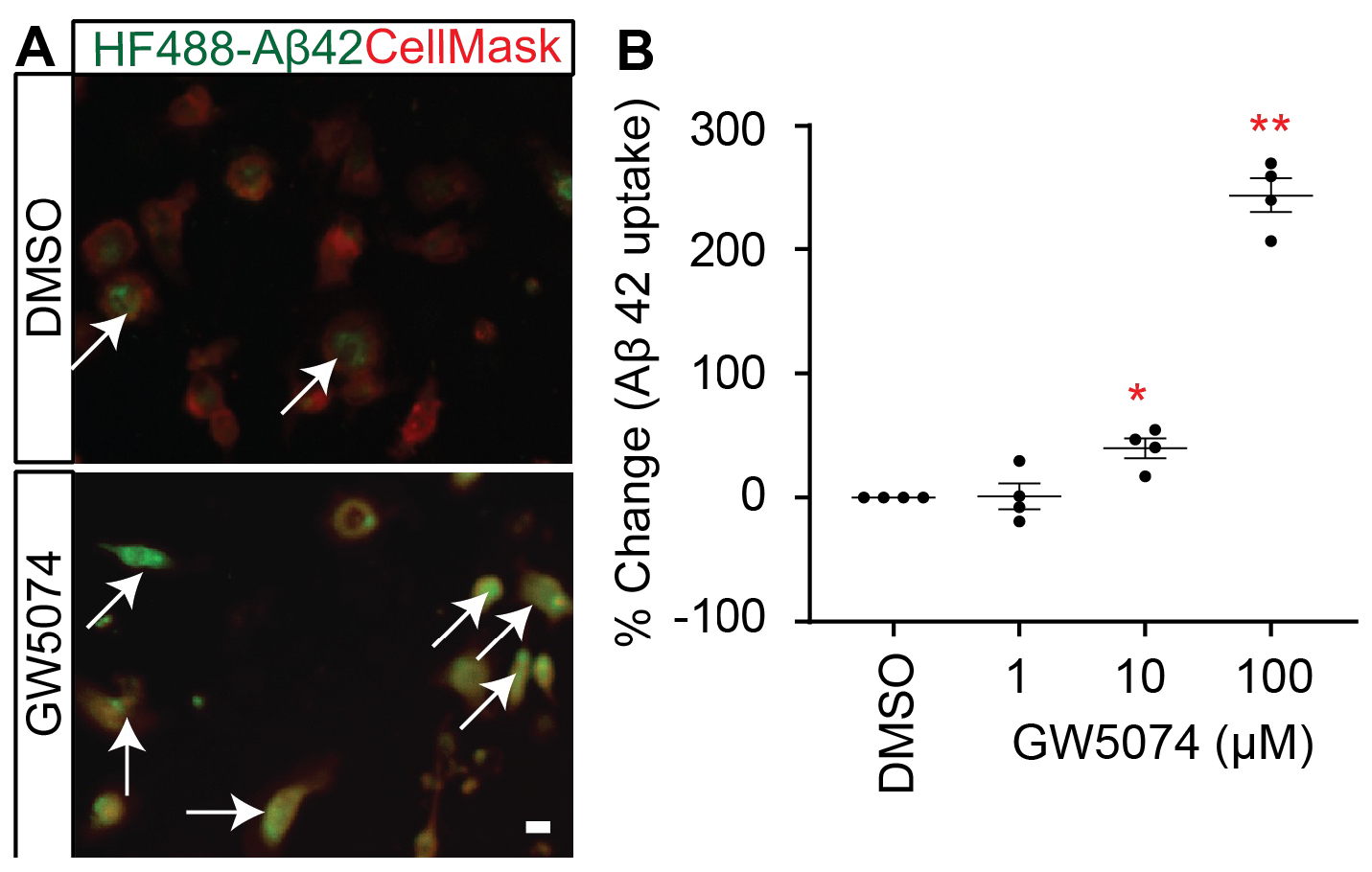
Figure 1.GW5074 optimization in MDMi with amyloid-β 1–42. (A) A representative image of MDMi treated with either DMSO or GW5074 and incubated with HyLite Fluor-488 conjugated Aβ1–42 for 2 h. CellMask was used to label the plasma membrane. Arrows indicate increased internalized Aβ in the GW5074 treated MDMi. (B) MDMi were treated with different concentrations of the drug for 24 h and subsequently incubated with HyLite Fluor-488 conjugated Aβ1–42 for 2 h. Cells were imaged on the Celigo Imaging Cytometer (Nexcelom Bioscience) and measured for live cell Aβ1–42 signal intensity. Data were analyzed using Student’s t-test. *p < 0.05, **p < 0.01. Scale bar, 10 μm. Each dot represents individual subjects.
As recently reported in Frontiers in Cellular Neuroscience, our screen identified several molecules which increase the uptake of Aβ1–42 by MDMi. Among the hits, GW5074 [IUPAC name: 3-(3,5-Dibromo-4-hydroxybenzyliden)-5-iodo-1,3-dihydroindol-2-one], a potent inhibitor of the c-RAF kinase, significantly increased the Aβ1–42 uptake ability of the MDMi. Notably, GW5074 was previously reported to be neuroprotective for cerebellar granule cells and cortical neurons. We found that GW5074 significantly increased the expression of key AD-associated microglial molecules known to modulate phagocytosis: TYROBP, SIRPβ1, and TREM2. Given that GW5074 is neuroprotective and blood-brain barrier permeable, the identified drug GW5074 is of interest potentiality from both neuronal and microglial perspectives—targeting key cell types rather than pathology.
Elizabeth M. Bradshaw, PhD
Adler Assistant Professor of Neurological Sciences (in Neurology, the Taub Institute for Research on Alzheimer's Disease and the Aging Brain, and the Institute for Genomic Medicine)
emb2280@cumc.columbia.edu
 |
 |  | |
| Jennifer S. Rabin, PhD | Emma Nichols | Adam M. Brickman, PhD |
Cerebral amyloid angiopathy (CAA), a form of cerebrovascular pathology resulting from the accumulation of β-amyloid (Aβ) in vessel walls, is thought to increase the risk of Alzheimer’s disease (AD) and is commonly found concurrent to AD in the aging brain. In the current study, together with co-first authors Dr. Jennifer Rabin (University of Toronto) and Emma Nichols (Johns Hopkins), we examined the impact of these two neuropathologies, CAA and neuritic Aβ plaques. Using data from autopsied subjects recruited from one of three longitudinal clinical-pathological cohort studies and causal statistical models, we first examined whether CAA influences tau deposition and cognitive decline independently or synergistically with Aβ burden. Secondly, we examined whether tau burden mediates the association between CAA and cognitive decline.
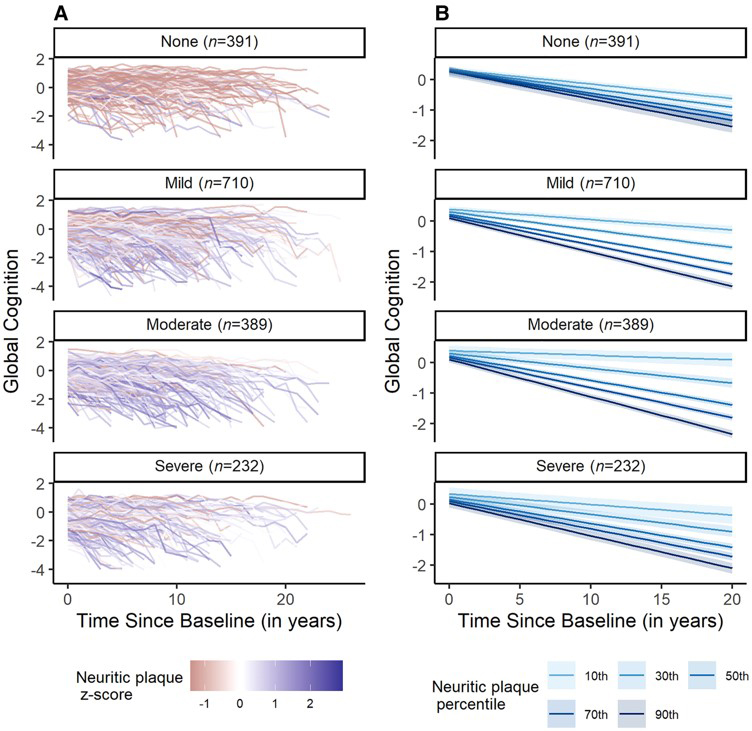
Figure 5. Cognitive trajectories by neuritic plaque burden and CAA severity. Both raw (A) and modelled (B) trajectories depict the significant interaction between neuritic plaque burden and CAA on cognitive decline. Cognitive decline was steepest in those with higher neuritic plaque burden and more severe CAA. Results are from a linear mixed-effects model controlling for age at baseline, gender, race, years of education, APOE4 status, baseline depressive symptoms, baseline vascular risk, number of visits and the interval between the last visit and death.
As recently reported in BRAIN with an accompanying editorial, we found that CAA interacted with neuritic plaques to accelerate tau burden and cognitive decline. Specifically, those with more severe cerebral amyloid angiopathy pathology and higher levels of neuritic plaque burden had greater tau burden and faster cognitive decline. We also found that tau mediated the association between cerebral amyloid angiopathy and cognitive decline among participants with higher neuritic plaque burden. In summary, more severe levels of CAA and higher parenchymal Aβ burden interacted to promote cognitive decline indirectly via tau deposition. These results highlight the dynamic interplay between cerebral amyloid angiopathy and Alzheimer’s disease pathology in accelerating progression toward dementia. These findings have implications for Alzheimer’s disease clinical trials and therapeutic development.
Adam M. Brickman, PhD
Professor of Neuropsychology (in Neurology, the Taub Institute for Research on Alzheimer's Disease and the Aging Brain and the Gertrude H. Sergievsky Center)
amb2139@cumc.columbia.edu
 | | |
| Patrick Lao, PhD | Adam M. Brickman, PhD |
Cerebrovascular disease is often excluded from studies of Alzheimer’s disease (AD) as a comorbidity, but it may play a mechanistic role in pathogenesis. Further, clinic-based studies often do not have the range of vascular burden to reliably assess vascular contributions to AD. In the current study, led by new faculty member Dr. Patrick Lao, we examined the relative contributions of neuroimaging-based biomarkers of amyloid, cerebrovascular disease, and neurodegeneration to cognitive decline over time within three major racial/ethnic groups from the Washington Heights-Inwood Columbia Aging Project (WHICAP).
As recently reported in Neurobiology of Aging, we found that 1) higher amyloid was associated with faster memory decline in all three racial/ethnic groups; 2) higher amyloid was further associated with faster non-memory decline in Non-Hispanic Black and Hispanic participants; 3) higher white matter hyperintensities (WMH), were associated with faster language, processing speed/executive function, and visuospatial ability decline in Non-Hispanic Black participants, while infarcts were associated with faster processing speed/executive function decline in Non-Hispanic White participants. In other words, amyloid may affect cognitive trajectories more broadly (i.e., decline in several domains beyond just memory) in minoritized racial/ethnic groups, potentially through the added influence of cerebrovascular disease; different forms of cerebrovascular disease (i.e., small vessel disease measured as WMH vs large vessel disease measured as infarcts) may affect cognitive trajectories uniquely; and cerebrovascular disease affects cognitive trajectories in all racial/ethnic groups when it is present, but it is often more present in minoritized racial/ethnic groups due to social determinants of health, including untreated heart disease.
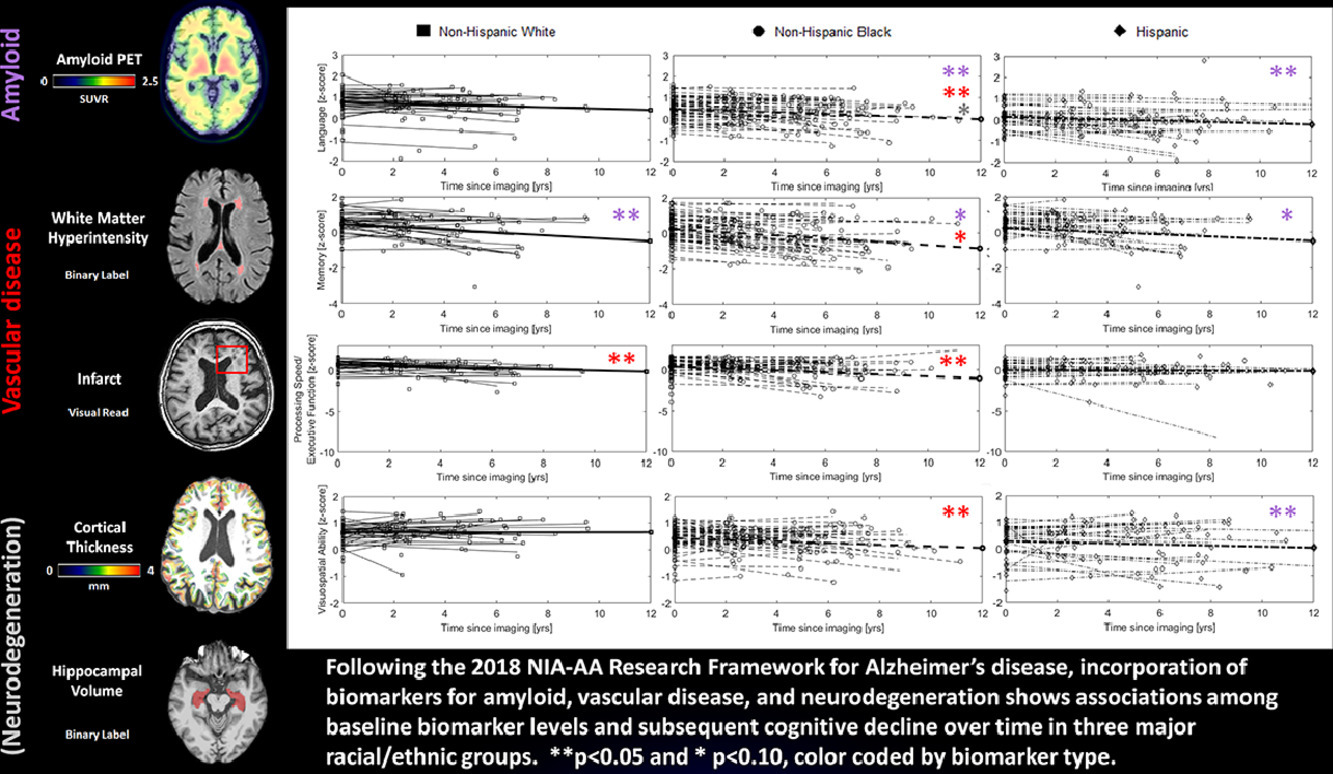 |
Overall, our study demonstrates the additive contribution of cerebrovascular biomarkers to the research framework for Alzheimer's disease established by the NIA, particularly when considering social determinants of health in minoritized racial/ethnic groups. Future work includes the incorporation of more recently available tau PET biomarkers, the use of plasma-based biomarkers, and the consideration of interactions among cerebrovascular and Alzheimer’s disease biomarkers.
Patrick Lao, PhD
Assistant Professor of Neurological Sciences (in Neurology and in the Gertrude H. Sergievsky Center)
pjl2133@cumc.columbia.edu
Adam M. Brickman, PhD
Professor of Neuropsychology (in Neurology, the Taub Institute for Research on Alzheimer’s disease and the Aging Brain, and the Gertrude H. Sergievsky Center)
amb2139@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.