
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspectives:
October 2021
1: An Immune Response Characterizes Early Alzheimer's Disease Pathology and Subjective Cognitive Impairment in Hydrocephalus Biopsies
2: MEF2C Common Genetic Variation Is Associated With Different Aspects of Cognition in Non-Hispanic White and Caribbean Hispanic Non-demented Older Adults
3: Association of Regional White Matter Hyperintensities With Longitudinal Alzheimer-Like Pattern of Neurodegeneration in Older Adults
4: Age of Onset of Huntington's Disease in Carriers of Reduced Penetrance Alleles
2: MEF2C Common Genetic Variation Is Associated With Different Aspects of Cognition in Non-Hispanic White and Caribbean Hispanic Non-demented Older Adults
3: Association of Regional White Matter Hyperintensities With Longitudinal Alzheimer-Like Pattern of Neurodegeneration in Older Adults
4: Age of Onset of Huntington's Disease in Carriers of Reduced Penetrance Alleles
 |  | |
| Wenrui Huang, PhD | Andrew Teich, MD, PhD |
Ongoing work molecularly characterizing Alzheimer’s disease (AD) autopsy brain tissue has produced a wealth of information about a wide range of pathophysiologic processes in AD. Less work has been done to molecularly characterize AD pathology in surgical biopsy tissue from living patients, which is more difficult to obtain but offers unique advantages for studying AD pathophysiology. Note that AD is not a surgical disease, and so biopsies that are studied in this way were not originally taken for AD, and AD pathology is incidentally discovered during pathologic examination. Surgical biopsies are free of gene expression changes that accompany end-of-life hypoxia/agonal state, as well as any changes in RNA caused by post-mortem degradation, and all cognitive and clinical data curated from the patients’ peri-operative charts is contemporaneous with the time of tissue acquisition. Thus, surgical biopsies with incidental AD pathology represent a unique opportunity to examine the pathophysiologic effects of AD.
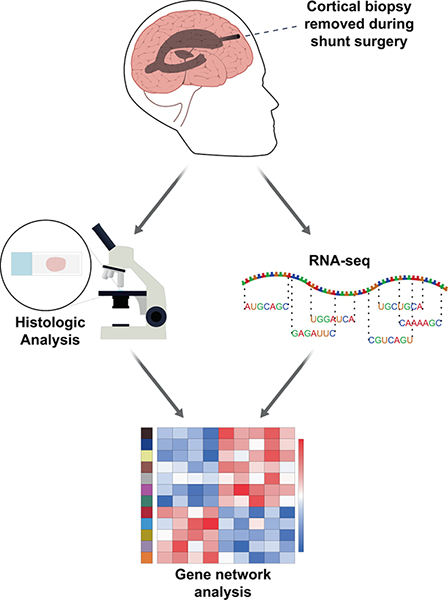
Figure 1. Biopsies removed for ventricular shunting in the operating room are immediately split in half. Half of the biopsy is frozen in liquid nitrogen and sent for RNA-seq. The other half is formalin fixed and paraffin embedded for subsequent pathologic analysis (see “Methods”).
One common source of biopsies with incidental AD pathology are from Normal Pressure Hydrocephalus (NPH) patients. NPH is a form of chronic hydrocephalus in the elderly, and placing a ventricular shunt is often effective for symptom relief. At the time of shunt placement, a cortical biopsy is often obtained at the brain entry point to look for possible coexistent brain pathology. Perhaps not surprisingly, cortical biopsies taken from elderly NPH patients at shunt placement have been shown to have a relatively high frequency of β-amyloid plaque pathology, ranging from 42% to 67%. The average age at biopsy for NPH patients (low to mid 70’s) is close to the average age of initial clinical presentation for AD (75.5), and AD pathology on biopsy is predictive of future development of clinical AD. All of this suggests that studying AD pathology in NPH biopsies may shed light on pathophysiologic changes associated with the early stages of AD.
In this work, recently published in Nature Communications, we report RNA-seq data from 106 NPH cortical biopsies and correlate changes in gene expression with co-morbid AD pathology. Analysis of these biopsies shows a restricted set of microglial and non-microglial genes that correlate with histological measurements of β-amyloid and tau pathology primarily in patients that report subjective cognitive impairment. Specifically, we identify a gene expression module enriched for murine disease-associated genes that positively correlates with AD pathology and a module enriched for murine homeostatic genes that negatively correlates with AD pathology, and in aggregate this is more consistent with the existing mouse literature than other publicly available AD autopsy datasets. Taken together, these data highlight the early microglial response to AD pathology in human cortical tissue, and may point to how this response is reproduced in AD mouse models.
Andrew F. Teich, MD, PhD
Assistant Professor of Pathology and Cell Biology (in Neurology)
Director, New York Brain Bank at Columbia University
aft25@cumc.columbia.edu
 |  | |
| Preeti Sunderaraman, PhD | Sandra Barral, PhD |
Genetic variants associated with late-onset Alzheimer’s disease (LOAD) risk can potentially be used as predictive biomarkers for dementia if these variants are also associated with cognition in healthy older adults prior to onset of cognitive impairment. For example, the apolipoprotein E (APOE), highly associated with LOAD risk, has been found to be strongly associated with memory in healthy adults before the clinical manifestation of AD. Myocyte Enhancer Factor 2C (MEF2C) has also been identified as a candidate gene contributing to the risk of developing AD, yet little is known about the role of MEF2C in specific aspects of cognition.

Table 1 presents baseline demographic and clinical characteristics of the participants.
Together with Taub colleagues Drs. Cosentino, Schupf, Manly, and Gu, our study is the first to investigate the association of common variants in the MEF2C gene and cognitive performance in four different domains, including memory, processing speed, visuospatial functioning and language. Participants from two ethnic groups, Non-Hispanic White (NHW; n=537) and Caribbean Hispanic (CH; n=1,197) from the Washington Heights-Hamilton Heights-Inwood Columbia Aging Project (WHICAP) study, were included. As recently reported in Frontiers in Genetics, single nucleotide polymorphisms (SNP) variants in the MEF2C gene showed nominally significant associations in all cognitive domains but for different SNPs across the NHW and CH ethnic groups.
Overall, our findings shed light on an apparent role for MEF2C in both memory and non-memory aspects of cognition, and highlight the differences in genetic architectural variations among those from different ancestries that should be considered in future studies.
Preeti Sunderaraman, PhD
Assistant Professor, Department of Neurology, Boston University School of Medicine
psun@bu.edu
Sandra Barral, PhD
Associate Professor of Neurogenetics (in Neurology, the Sergievsky Center, and the Taub Institute) at CUIMC
smb2174@cumc.columbia.edu
 |  |  | ||
| Batool Rizvi, MS | Patrick J. Lao, PhD | Adam M. Brickman, PhD |
Small vessel cerebrovascular disease, visualized as white matter hyperintensities (WMH), is associated with cognitive decline and risk of clinical Alzheimer’s disease (AD), but it is unclear how or why. Previous cross-sectional work led by our laboratory and others suggests that small vessel cerebrovascular disease may promote neurodegenerative changes related to AD, which may have particular relevance to racially and ethnically minoritized groups. Building on this research, together with first author Batool Rizvi and colleagues from Taub, including second-author Dr. Patrick J. Lao, our latest study examined whether total and regional WMH volumes were associated with a pattern of longitudinal cortical thinning that is anatomically similar to what is typically seen in AD among 303 racially and ethnically diverse older adults from the WHICAP study.
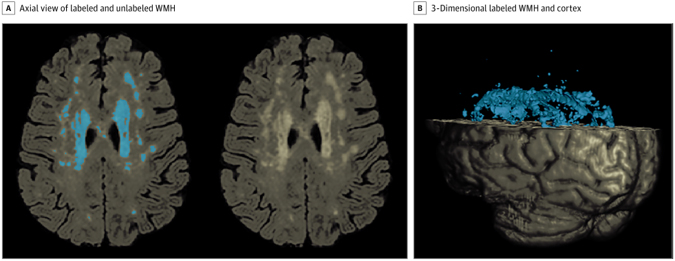
Figure 1. White Matter Hyperintensity (WMH) Quantification A, Axial view of a fluid-attenuated inversion recovery brain extracted image demonstrating labeled WMH with in-house developed software on the left and unlabeled WMH distribution on the right. B, A 3-dimensional rendering of labeled WMH and cortex.
As recently reported in JAMA Network Open, we found that baseline WMH volumes were associated with cortical thinning in medial temporal and frontal/parietal regions. Specifically, total WMH volume was associated with cortical thinning in the right caudal middle frontal cortex and paracentral cortex, whereas parietal WMH volume was associated with atrophy in the left entorhinal cortex and right rostral middle frontal, paracentral, and pars triangularis cortices. Thinning of the right caudal middle frontal and left entorhinal cortices was related to lower scores on a memory test. Many of the associations observed between total and regional WMH volume and cortical thinning were stronger in non-Hispanic Black participants compared with White participants.
In conclusion, our work suggests that small vessel cerebrovascular disease drives neurodegenerative changes, which overlap anatomically with patterns we typically see in Alzheimer’s disease, and subsequent memory decline. Many of this observations were stronger among Black older adults, which may help explain some of the well-documented race differences in incidence and prevalence of clinical AD. Overall, these new findings provide support for the hypothesis that cerebrovascular disease affects risk and progression of AD by promoting neurodegeneration and memory decline.
Batool Rizvi, MS
Neurobiology and Behavior PhD student at UC Irvine
brizvi@uci.edu
Adam M. Brickman, PhD
Professor of Neuropsychology (in Neurology, the Taub Institute, and the Sergievsky Center)
amb2139@cumc.columbia.edu
Age of Onset of Huntington's Disease in Carriers of Reduced Penetrance Alleles
 |  | |
| Erin McDonnell, PhD | Yuanjia Wang, PhD | |
 |  | |
| Jill S. Goldman, MS, MPhil, CGC | Karen Marder, MD, MPH |
Huntington's disease (HD) is a fatal autosomal dominant neurodegenerative disorder caused by a CAG repeat expansion in the huntingtin gene (HTT). Age of onset in HD is believed to correlate with the number of HTT CAG repeats. For those with 40 or more CAG repeats that survive to adulthood, penetrance is 100%, yet less is known about age of onset for those with a “reduced penetrance (RP)” range of 36 to 39 repeats. Thus, leveraging the most recent data from Enroll-HD, the largest longitudinal observational study for HD, we sought to provide empirical estimates of HD penetrance over time for each allele within the RP range.
Together with lead author Dr. Erin McDonnell (Mailman Biostatistics PhD ‘21), we analyzed 431 pre-manifest RP allele carriers. Cumulative penetrance (CP) was estimated from Kaplan–Meier curves. As recently reported in Movement Disorders, no one with 36 repeats (n=25) reached manifest HD. CP for 38 repeats (n=120) was 32% and 51% by ages 70 and 75, respectively, and 68% and 81% by ages 70 and 75 for 39 repeats (n=253). CP was not estimable for 37 repeats (n=33).
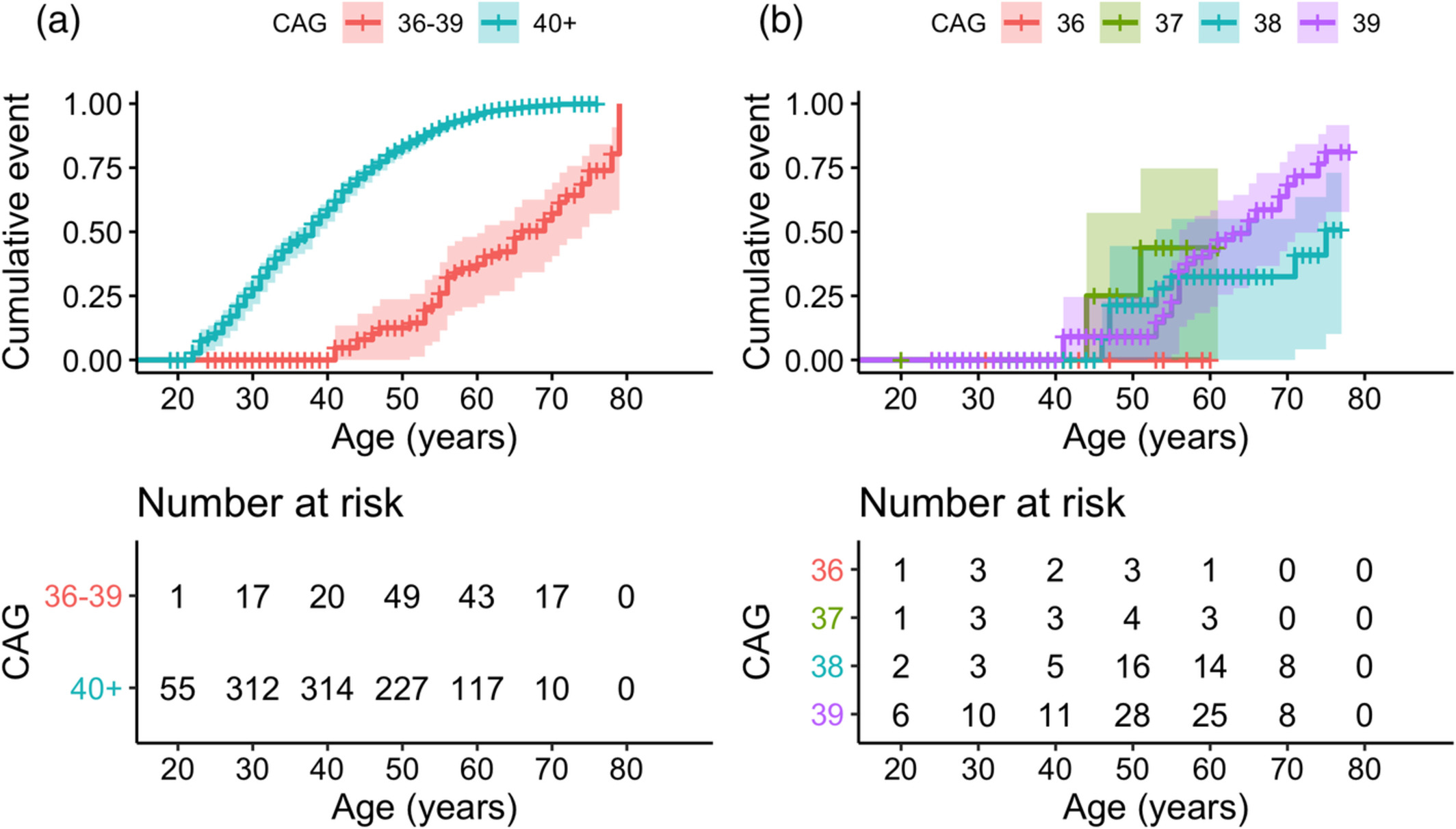
Figure 1. Cumulative penetrance over age, stratified by CAG repeat length. (a) Stratified by CAG repeat length 36–39 versus 40+. Log‐rank P value <0.001. (b) Individuals with RP alleles only, stratified by individual repeat length. Log‐rank P value = 0.1. Comparing only 38 versus 39 repeats: P value = 0.07. Kaplan–Meier curves were administratively censored at the earliest age beyond 60 years that the risk set contained only 1 individual.
This analysis is clinically important for genetic counseling of individuals from known HD families with repeat lengths in the RP range who want more definitive information of their risk of phenoconversion. It is also important for the general population. One study estimated that 1 in 400 people in the general population have a CAG repeat expansion ≥36, most of which are in the RP range.
Karen Marder, MD, MPH
Sally Kerlin Professor of Neurology (in the Sergievsky Center, Taub Institute, and Psychiatry)
ksm1@cumc.columbia.edu
Jill S. Goldman, MS, MPhil, CGC
Professor of Genetic Counseling (in Neurology) at CUIMC
jg2673@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.