
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspectives:
November 2021
1: KYNA/Ahr Signaling Suppresses Neural Stem Cell Plasticity and Neurogenesis in Adult Zebrafish Model of Alzheimer's Disease
2: Characterization of Mitochondrial DNA Quantity and Quality in the Human Aged and Alzheimer's Disease Brain
3: Self-Awareness for Financial Decision Making Abilities is Linked to Right Temporal Cortical Thickness in Older Adults
2: Characterization of Mitochondrial DNA Quantity and Quality in the Human Aged and Alzheimer's Disease Brain
3: Self-Awareness for Financial Decision Making Abilities is Linked to Right Temporal Cortical Thickness in Older Adults
 |  |  | ||
| Richard Mayeux, MD, MSc | Giuseppe Tosto, MD, PhD | Caghan Kizil, PhD |
Adult neurogenesis and neuroregeneration capacity evolutionarily vary within the vertebrate phylogeny. Aquatic vertebrates such as teleost zebrafish bear a life-long neurogenic ability throughout their entire brain, while mammals have restricted stem cell niches and neurogenic outcome. Additionally, zebrafish can produce new functional neurons on demand upon injury or disease, while mammalian brains fail to do so. This disparity translates into significantly different neuroregenerative abilities in fishes and mammals and begs the question whether zebrafish can teach us how to efficiently regenerate new neurons to counteract the detrimental outcomes of neurodegeneration or to enhance the resilience of the brain during aging.
Neural stem/progenitor cells are of astroglial lineage and are the main reservoir for new neurons to be made. The molecular cues that regulate the neurogenic ability in these cells are not fully known, but constitute a fertile research area that is clinically relevant. Many neurological diseases have an etiological component that might be related to altered neurogenic outcome. Recent studies showed significant reduction in neurogenesis in the human brains during AD. Reduced neurogenesis could be a contributing factor to the disease, and counteracting the waning neurogenesis might be a disease modifying intervention by enhancing the resilience of the brain. To understand the molecular mechanisms that enable successful neuroregeneration and those that could be harnessed for novel clinical therapeutic directions, we are aiming to provide a deeper understanding of the neuroregenerative mechanisms in adult zebrafish brain model of neurodegeneration.
In our study, published recently in Cells, we identified a new subset of astroglial cells in adult zebrafish brain that are normally kept quiescent but are activated to form neurons after amyloid toxicity. These cells are responsive to a specific tryptophan metabolite, kynurenic acid (KYNA). We found that KYNA negatively regulates neurogenesis in zebrafish brain through its receptor, aryl hydrocarbon receptor 2 (Ahr2) during the homeostatic conditions. KYNA is produced by periventricular neurons that are juxtaposed to the astroglia and express the key enzyme KAT2, which converts tryptophan to kynurenic acid. The expression levels of KAT2 reduces after amyloid toxicity in the zebrafish brain and this allows the Ahr2-expressing neural stem cells to proliferate and form new neurons. Zebrafish brain suppresses Ahr signaling to enable pathology-induced neurogenesis. Ahr2 expression defines a subset of neural stem cells that are distinct from the two types that we previously identified, which are marked by expression and functional relevance of Interleukin-4 receptor (Il4r) and Nerve growth factor receptor (Ngfr). Ahr2-positive cells, therefore, represent a new type of quiescent neural progenitors that respond to AD pathology.
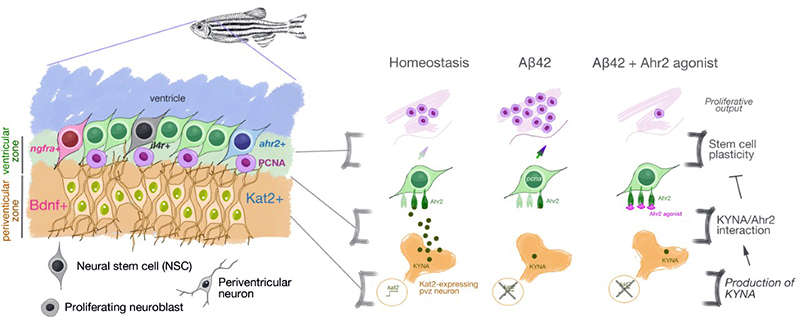 Figure 1. (Left) KYNA/Ahr2 signaling defines the third distinct Alzheimer’s disease-responsive subset of neural stem cell population in adult zebrafish brain in addition to Interleukin-4 and Nerve growth factor-responsive neural stem cells; (Right) Schematic view of negative regulation of neural stem cell plasticity and neurogenesis in adult zebrafish brain by KYNA/Ahr2 signaling. Aβ42 reduces the expression levels of KAT2, which generates KYNA, an inhibitory regulator of neural stem cell proliferation through its receptor Ahr2.
|
To compare the findings to human brains with AD, we performed transcriptome-wide association study and reported several genes involved in Ahr signaling and KYNA production were differentially expressed in late-onset Alzheimer’s disease patients when compared to healthy individuals. Confirming our previous findings, we also found KAT2 enzyme levels are significantly increased in AD patient brains, further strengthening the link between defective neurogenesis and AD. Ahr signalling is well-known for linking the environmental toxicity to gene regulation in humans, and our study also proposes a connection between environmental effects to regulation of neurogenesis.
Caghan Kizil, PhD
Visiting Professor of Neurological Sciences (in Neurology)
ck2893@cumc.columbia.edu
 |
 |
|
| Hans-Ulrich Klein, PhD | Philip L. De Jager, MD, PhD |
Mitochondrial dysfunction is a feature of neurodegenerative diseases, including Alzheimer’s disease (AD). Changes in the mitochondrial DNA copy number (mtDNAcn) and increased mitochondrial DNA mutation burden have both been associated with neurodegenerative diseases and cognitive decline. Together with colleagues including Drs. Caroline Trumpff and Martin Picard from the Departments of Neurology and Psychiatry, we quantified mtDNAcn and small heteroplasmic mtDNA mutations in n = 1361 well-characterized human brain samples from 5 different regions using whole-genome sequencing to systematically identify which common brain pathologies are associated with mitochondrial recalibrations and to disentangle the relationship between these pathologies, mtDNAcn, mtDNA heteroplasmy, aging, neuronal loss, and cognitive function.
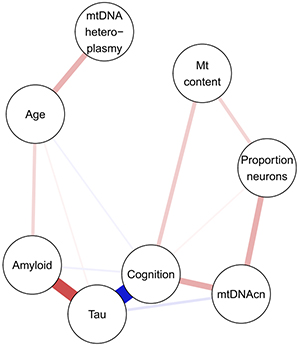
Figure 1. Partial correlations between mitochondrial, pathologic and cognitive measures in the DLPFC. Positive associations are depicted in red, negative associations in blue.
As recently reported in Molecular Neurodegeneration, mtDNAcn was 7–14% lower in AD relative to control participants. When accounting for all 10 common neuropathologies, only tau was significantly associated with lower mtDNAcn in the dorsolateral prefrontal cortex (DLPFC). In the posterior cingulate cortex, TDP-43 pathology demonstrated a distinct association with mtDNAcn. No changes were observed in the cerebellum, which is affected late by pathologies. Neither age nor gender was associated with mtDNAcn in the studied brain regions when adjusting for pathologies. Mitochondrial content and mtDNAcn independently explained variance in cognitive function unaccounted by pathologies, implicating complex mitochondrial recalibrations in cognitive decline. In contrast, mtDNA heteroplasmy levels increased by 1.5% per year of life in the cortical regions, but displayed no association with any of the pathologies or cognitive function (Figure 1).
In summary, we showed that tau and not amyloid-β is primarily associated with reduced mtDNAcn. In the posterior cingulate cortex, the association of TDP-43 with low mtDNAcn points to a vulnerability of this region in limbic-predominant age-related TDP-43 encephalopathy. While we found low mtDNAcn in brain regions affected by pathologies, the absence of associations with mtDNA heteroplasmy burden indicates that mtDNA point mutations and small indels are unlikely to be involved in the pathogenesis of late-onset neurodegenerative diseases.
Hans-Ulrich Klein, PhD
Assistant Professor of Neurological Sciences (in Neurology and the Taub Institute)
hk2948@cumc.columbia.edu
Philip L. De Jager, MD, PhD
Weil-Granat Professor of Neurology (in Neurology and the Taub Institute)
pld2115@cumc.columbia.edu
 |  | |
| Preeti Sunderaraman, PhD | Stephanie Cosentino, PhD |
Older adults are at risk for suboptimal financial decision making (FDM) and adverse consequences associated with such, including making poor investments and being at increased risk for financial exploitation. Awareness of the integrity of one’s own FDM abilities (or financial awareness) is likely a critical construct to define and measure in order to identify those at risk for making suboptimal financial decisions. In the current study, we examined both the cognitive and cortical thickness correlates of financial awareness in 59 community-dwelling older adults. FDM was measured objectively using the Financial Competence Assessment Inventory, which includes a range of items from writing a check to defining financial terms. Financial awareness was also measured objectively by asking participants to make judgments about their ability to answer each question correctly. Awareness was operationalized as the average discrepancy between predictions and performance, with positive discrepancies reflecting overconfidence and negative discrepancies reflecting underconfidence. We hypothesized that while FDM performance itself would correlate with cognitive abilities, such as numeracy and executive function, we anticipated that financial awareness would be linked more closely to metacognitive abilities including memory awareness, and cortical thickness in brain regions previously shown to support self-evaluative processes more broadly.
Figure 2. The Social Cognitive Neuroscience model of financial exploitation risk (Spreng, Karlawish & Marson, 2016; reproduced with permission). Panel A depicts the original proposed model, whereas panel B shows one possible mechanism by which self-awareness may contribute to the model. In this figure, awareness may moderate the pathways associated with financial exploitation risk while the brain function and structure may mediate the associations among the pathways and financial exploitation risk.
As recently reported in Brain Imaging and Behavior, we found that, as hypothesized, in contrast to FDM itself, financial awareness was linked to memory awareness. Moreover, both of these metacognitive abilities were associated with right-hemisphere temporal thickness, underscoring the idea that a common network contributes to self-evaluative processes across a range of abilities. Interestingly, studies on financial exploitation find the right temporal cortex to be associated with increased scam susceptibility. A recent study found that in non-demented adults, 3 clusters including the right-sided parahippocampal/hippocampal/fusiform, hippocampal, and middle temporal regions were inversely associated with scam susceptibility (Han et al., 2016). Based on these findings, one can speculate that compromised self-awareness, and financial awareness in particular, may be an important mechanism by which vulnerability to exploitation arises (Lichtenberg, Campbell, Hall, & Gross, 2020; Shao, Zhang, Ren, Li, & Lin, 2019; Spreng, Karlawish, & Marson, 2016). The Social Cognitive Neuroscience model offers a novel framework to understand the factors that feed an individual’s risk of financial exploitation (Spreng et al., 2016) including both cognitive and social capacities. It is conceivable that self-awareness may contribute to this framework primarily as a moderating variable and can affect the relationship between both or either pathway (social and/or cognitive) and exploitation risk (see Figure 2). Specifically, the moderating effect of self-awareness would be that higher self-awareness would reduce the effect of cognitive or social impairment on financial exploitation risk. Incorporating the contributing role of self-awareness in various models of financial exploitation will be an important consideration for future studies.
Preeti Sunderaraman, PhD
Former NRSA (F32) Postdoctoral Research Fellow (now at Boston University)
psun@bu.edu
Stephanie Cosentino, PhD
Associate Professor of Neuropsychology (in Neurology, the Gertrude H. Sergievsky Center, and the Taub Institute)
sc2460@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.