
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspective:
November 2024
1: ABCA7-Dependent Induction of Neuropeptide Y is Required for Synaptic Resilience in Alzheimer’s Disease Through BDNF/NGFR Signaling
2: Regulation of Synapse Density by Pumilio RNA-Binding Proteins
3: CD33 and SHP-1/PTPN6 Interaction in Alzheimer's Disease
4: A Neural Implementation of Cognitive Reserve: Insights from a Longitudinal fMRI Study of Set-Switching in Aging
2: Regulation of Synapse Density by Pumilio RNA-Binding Proteins
3: CD33 and SHP-1/PTPN6 Interaction in Alzheimer's Disease
4: A Neural Implementation of Cognitive Reserve: Insights from a Longitudinal fMRI Study of Set-Switching in Aging
 |  |  | ||
| Hüseyin Tayran | Elanur Yilmaz, PhD | Prabesh Bhattarai, PhD | ||
 |  | |||
| Richard Mayeux, MD, MSc | Caghan Kizil, PhD |
In our recent publication in Cell Genomics, we explored the critical role of the ABCA7 gene in mediating synaptic resilience through the regulation of neuropeptide Y (NPY) in Alzheimer’s disease (AD). Using a multidisciplinary approach, including a CRISPR-Cas9-mediated zebrafish model, patient-derived iPSCs, transcriptomics, and clinical patient data, we identified a mechanism how ABCA7 regulates brain resilience in Alzheimer's disease. Our zebrafish model of amyloid-beta (Aβ) toxicity revealed that ABCA7 is indispensable for maintaining and inducing the expression of Neuropeptide Y, which plays a pivotal role in the brain’s resilience against AD pathology. Single-cell transcriptomics demonstrated that the loss of ABCA7 disrupts the NPY–BDNF–NGFR signaling axis, leading to reduced synaptic density, impaired astroglial proliferation, and a compromised resilience response. These findings were corroborated in patient-derived iPSCs, where ABCA7 knockout resulted in diminished NPY expression under Aβ-induced stress.
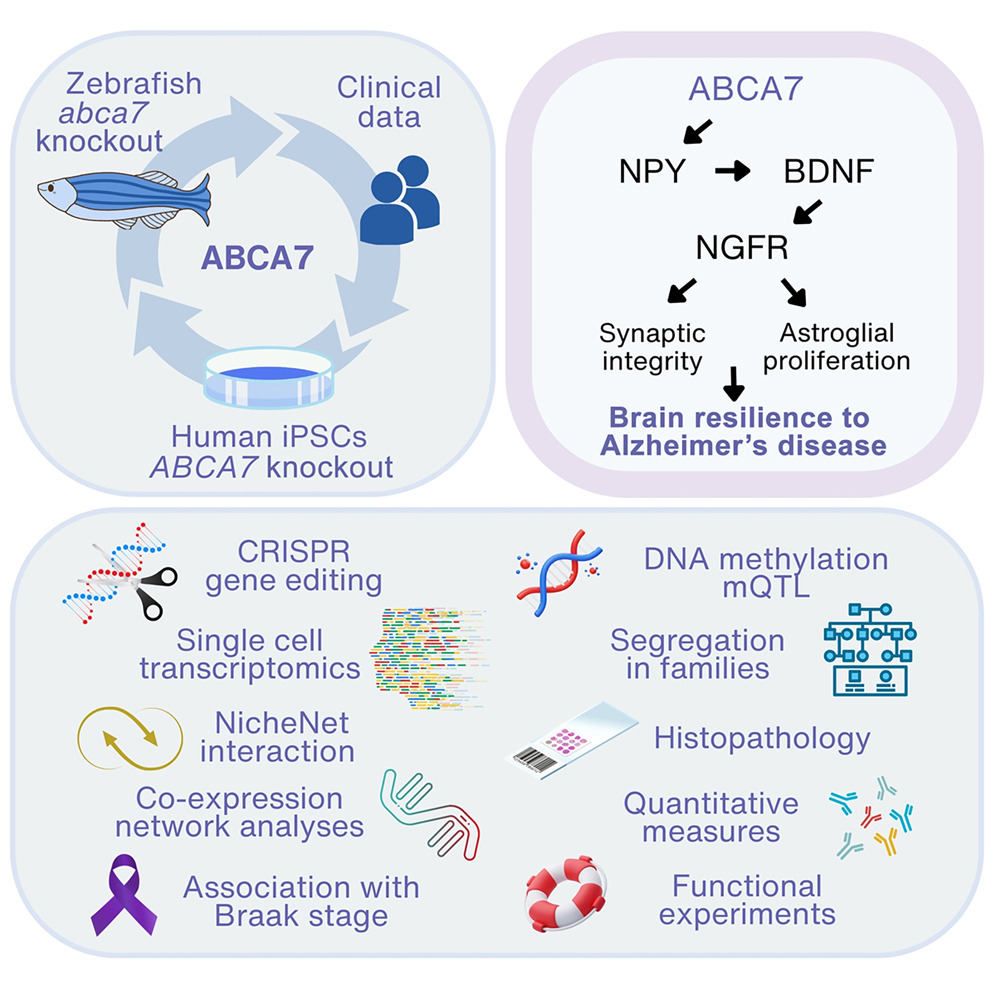
Graphical Abstract
A central finding of our work is the bidirectional relationship between NPY and BDNF. We showed that reduced NPY levels in ABCA7-deficient zebrafish and human neurons resulted in downstream suppression of BDNF expression, a neurotrophic factor essential for synaptic maintenance and neural plasticity. Rescue experiments demonstrated that ectopic NPY administration restored synaptic density and BDNF levels, emphasizing NPY's upstream regulatory role. As highlighted in the CUIMC Newsroom, clinical data from human cohorts confirmed the translational relevance of these findings. Reduced NPY levels in AD patients strongly correlated with advanced Braak stages, while epigenetic analyses linked ABCA7 variants to altered methylation patterns in NPY and BDNF promoters. This interplay underscores ABCA7's role in modulating epigenetic and transcriptional networks that govern brain resilience.
Our study also identified NGFR as a mediator of BDNF signaling in astroglia, a finding with significant implications for neurogenesis. In the zebrafish model, NGFR expression was tightly linked to astroglial proliferation, a process disrupted by ABCA7 loss. These insights align with our previous work demonstrating that NGFR activation in mammalian models enhances neurogenesis and mitigates AD pathology.
These findings position the ABCA7–NPY–BDNF–NGFR axis as a central mechanism underlying synaptic resilience and offer a promising therapeutic target. By leveraging NPY and BDNF signaling pathways, future interventions could amplify the brain's natural protective responses to AD-related pathology. This study exemplifies the power of integrative approaches, combining cross-species models with human-derived data, to unravel complex neurodegenerative mechanisms. By advancing our understanding of ABCA7's role in synaptic and cellular resilience, we contribute toward designing targeted therapies aimed at mitigating AD progression.
Caghan Kizil, PhD
Associate Professor of Neurological Sciences (in Neurology and in the Taub Institute)
ck2893@cumc.columbia.edu
Regulation of Synapse Density by Pumilio RNA-Binding Proteins
 |
 |
| Lisa Randolph, PhD | Ulrich Hengst, PhD |
Neurons are among the most morphologically complex, dynamic, and long-lived cells in the human body, leading to special challenges for the spatial and temporal control of gene expression. All aspects of neuronal development and function crucially depend on post-transcriptional mechanism for the control of gene expression, and essentially all these mechanisms involve the function of RNA-binding proteins (RBPs) that control among others a transcript’s splicing, localization, translation, and stability. Coordinated RNA regulation is crucial for example for axon growth and pathfinding, and at later developmental stages for synapse formation and stabilization.
Previously, we had identified that elevated expression levels of mammalian RNA-binding proteins Pumilio 2 (Pum2) during early developmental stages retained mRNAs, including transcripts encoding synaptic proteins within the cell body, thereby preventing their localization and translation in axons and dendrites. Given that Pum2 and its homologue Pum1 are developmentally downregulated around the time of synapse formation, we hypothesized that reduced expressions of Pum2 and potentially Pum1 would constitute an endogenous mechanism for the precise timing of synapse formation.
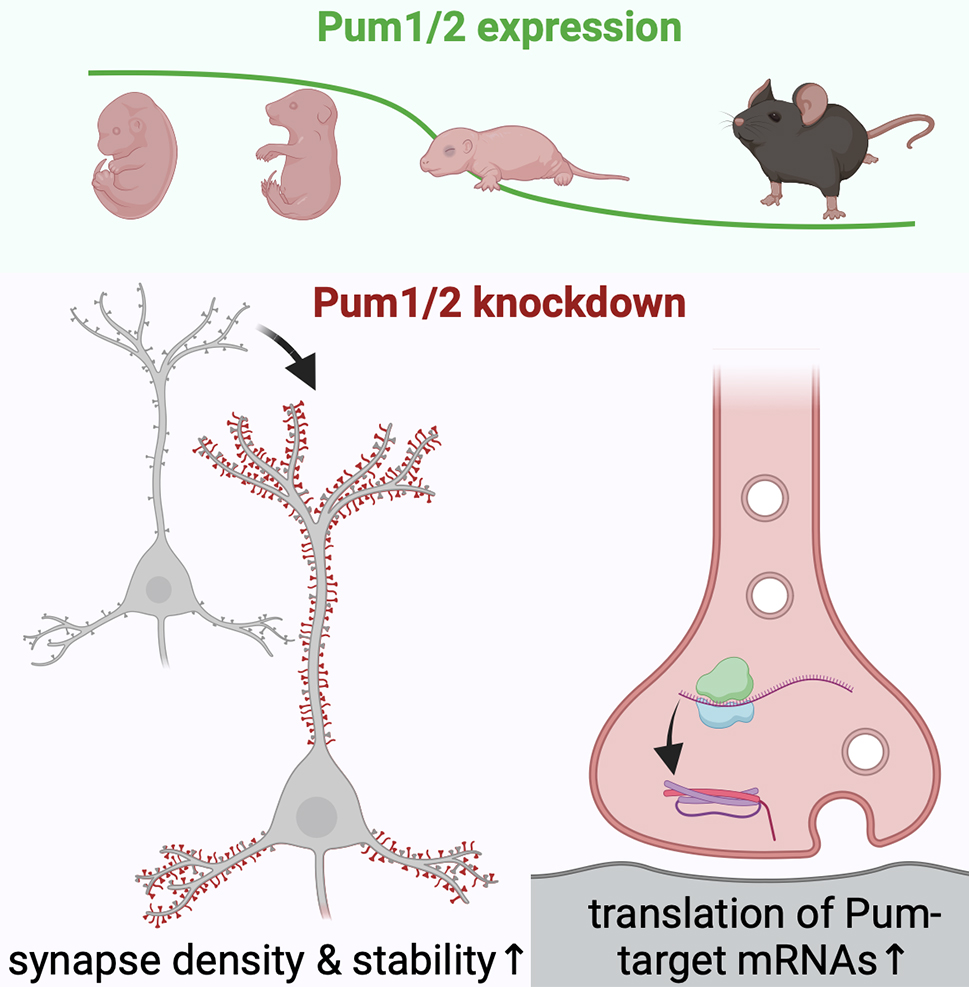
Figure: Pumilio RNA-binding proteins exhibit developmental downregulation in the brain. Decreased expression of Pum1/2 facilitates synapse maturation and enhances the density of excitatory and inhibitory synapses. The regulation of Pumilio levels constitutes an intrinsic cellular mechanism for modulating synapse formation.
In our recently published Cell Reports study, Dr. Lisa Randolph, then a graduate student in the Neurobiology and Behavior Program at Columbia University, tested this hypothesis by prematurely suppressing the expression of Pum1 and 2 in cultured hippocampal and cortical neurons. Indeed, simultaneous knockdown of the Pumilios resulted in significantly increased synapse density. Contrary to our expectations, the augmentation in synapse numbers was not attributable to earlier synapse formation but rather to enhanced maturation of newly formed synapses. In accordance with a potential function in controlling protein synthesis in the neuronal periphery, we detected elevated protein synthesis at synaptic sites upon Pum1/2 knockdown.
We propose a mechanism whereby Pumilio proteins repress the local synthesis of synaptic proteins, such as SNAP-25, thereby curbing the capacity of neurons to form and maintain synapses. Notably, this regulatory mechanism is operative not only during development but also after the majority of synapses have already been formed, as knockdown of Pum1/2 at later times still elicited exuberant synapse formation. Our findings have potential implications for future studies related to normal aging, as Pum2 is upregulated in late life, and its hyperactivity has been associated with an aging phenotype in model organisms.
Ulrich Hengst, PhD
Professor of Pathology and Cell Biology (in the Taub Institute for Research on Alzheimer’s Disease and the Aging Brain)
uh2112@cumc.columbia.edu
CD33 and SHP-1/PTPN6 Interaction in Alzheimer's Disease
 |  | |
| Mamunur Rashid, PhD | Annie J. Lee, PhD | |

|  | |
| Badri N. Vardarajan, PhD, MS | Elizabeth M. Bradshaw, PhD |
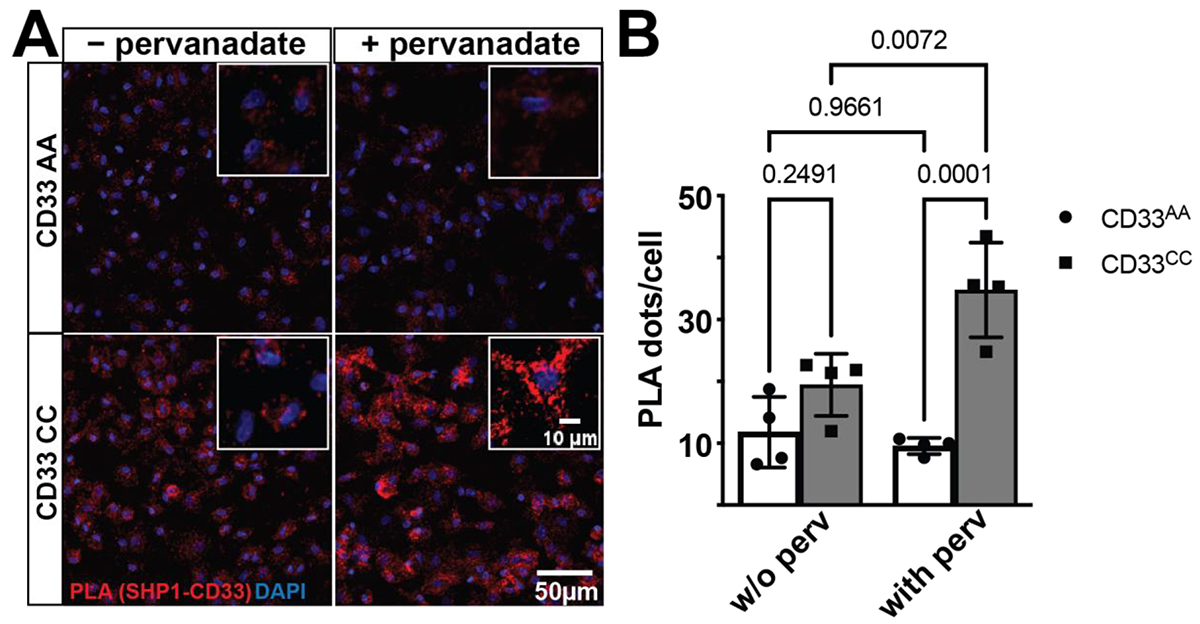
Figure: CD33 and SHP-1 interact in situ in a CD33 genotype-dependent manner. (A) A representative confocal image of post-mortem human prefrontal cortex stained for proximity ligation assays (PLA). The proximity ligation puncta (red dots, zoomed in inset) represent the protein–protein interaction between CD33 and SHP-1. DAPI (blue) is used to counterstain the nucleus. (B) Quantification of the PLA dots show a significant increase in the PLA counts in the CD33 CC group (n = 16 tissue sections, 11.36 ± 1.956) when compared to the CD33 AA groups (n = 10 tissue sections, 0.6185 ± 0.1482). Data represented as the mean ± SEM. *** p < 0.001, unpaired t-test. Scale bar: 100 μm; 10 μm inset.
CD33, a transmembrane glycoprotein expressed on myeloid cells, plays a crucial role in regulating immune responses through its inhibitory signaling motifs. Genetic variations in CD33, such as the rs3865444C risk allele, have been associated with Alzheimer’s disease (AD) susceptibility, influencing amyloid β clearance and contributing to plaque accumulation. SHP-1 (encoded by PTPN6) is a protein tyrosine phosphatase that binds to CD33’s ITIM motifs, modulating signaling pathways critical to microglial function. Understanding the interaction between CD33 and SHP-1 is essential for determining their role in AD pathophysiology and their potential as therapeutic targets.
As recently published in Genes, in collaboration with Dr. David Bennett (Rush University), we examined the critical interaction between CD33 and SHP-1 in the context of AD-associated genetic variation. We explored how genetic variations at the CD33 locus influence its binding with SHP-1, a key protein tyrosine phosphatase, using the proximity ligation technique in human microglia and microglia-like cells. Our findings demonstrate a genotype and activation state-dependent interaction between CD33 and SHP-1, which may contribute to the increased AD risk linked to certain CD33 variants. Moreover, we revealed that the interaction between CD33 and PTPN6 (SHP-1) significantly associates with AD-related pathological traits, while interactions with PTPN11 (SHP-2), a related protein tyrosine phosphatase that also binds CD33, do not show similar effects. These results emphasize the role of innate immune mechanisms in AD susceptibility and suggest new avenues for therapeutic interventions targeting these pathways.
Elizabeth M. Bradshaw, PhD
Adler Assistant Professor of Neurological Sciences (in Neurology and the Taub Institute)
Co-Director, The Carol and Gene Ludwig Center for Research on Neurodegeneration
emb2280@cumc.columbia.edu
 |  | |
| Fatemeh Hasanzadeh, PhD | Christian Habeck, PhD | |

|  | |
| Yunglin Gazes, PhD | Yaakov Stern, PhD |
In this longitudinal study spanning five years, we investigated how task-related neural activation during an experimental task-switching paradigm serves as a neural marker of cognitive reserve (CR). Specifically, our team, including postdoctoral scientist Dr. Fatemeh Hasanzadeh, as well as Drs. Christian Habeck and Yunglin Gazes, examined whether greater engagement of functional brain networks during an executive control function task could buffer cognitive performance against age-related structural brain changes that typically impair cognition.
In this executive task, participants see one letter at a time on the screen and must make a decision about it using a button press. If the letter is red, they make an lower/upper case decision; green letters require a vowel/consonant decision. In the “single-task” condition, only one color is presented throughout, and participants make the same decision for every letter. In the “dual-task,” the color of the letters changes throughout the trial and the participant must switch their response based on that color. "Switch cost" is the added time and reduced accuracy associated with dual- vs single-task.
We applied ordinal trend canonical variates analysis (OrT CVA) to functional MRI (fMRI) data from single and dual-task conditions. This advanced multivariate technique identified brain regions showing consistent activation changes as task difficulty increased from the single to dual task, characterizing individual differences in neural responses to task-switching demands. Concurrently, we constructed a multivariate measure of brain structure—combining cortical and subcortical volume and thickness changes—through elastic net regression, summarizing the structural brain changes that were associated with decline in task performance over 5 years.
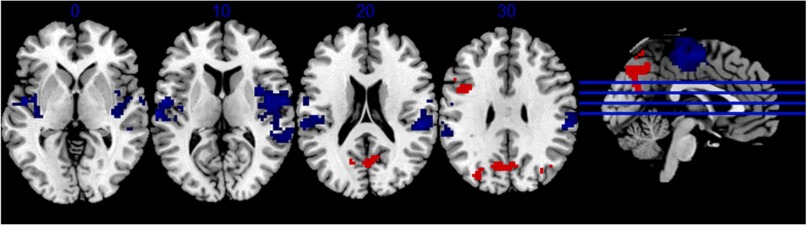
Figure: Areas of the brain that show concomitant increased (red) and decreased (blue) activation when switching from single to dual conditions. Participants who exhibited a greater utilization of this pattern at baseline showed greater resilience, and could lessen the effect of reduced brain reserve on cognitive performance.
Our results, recently published in the Neurobiology of Aging, revealed that the degree of differential activation between single and dual conditions observed at the initial visit moderated the relationship between structural brain changes and cognitive decline. Individuals with greater task-related activation exhibited a reduced impact of structural brain decline on switch cost, supporting the cognitive reserve theory that CR is a “property of the brain” that enables resilience against aging-related brain changes.
By moving beyond traditional CR proxies like IQ and education, this study provides direct evidence that differential functional activation can represent a neural implementation of CR. The findings highlight the critical role of differential utilization of a brain network in preserving cognitive flexibility, offering new insights into the mechanisms underlying cognitive reserve and healthy aging.
Yaakov Stern, PhD
Florence Irving Professor of Neuropsychology (in Neurology, Psychiatry, the Sergievsky Center, and the Taub Institute)
ys11@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.