
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspectives:
July 2017
#1 Abnormal Neurofilament Inclusions and Segregations in Dorsal Root Ganglia of A Charcot-Marie-Tooth Type 2E Mouse Model
#2 LTP and Memory Impairment Caused by Extracellular Aβ and Tau Oligomers is APP-Dependent
#2 LTP and Memory Impairment Caused by Extracellular Aβ and Tau Oligomers is APP-Dependent
View Archive
» #2 Neuropathologic Features of TOMM40 '523 Variant on Late-Life Cognitive Decline
» #2 An Approach to Studying the Neural Correlates of Reserve
» #1 Brain Atrophy Can Introduce Age-Related Differences in BOLD Response
» #2 Age-Related Biomarkers in LLFS Families With Exceptional Cognitive Abilities
» #2 Polygenic Risk Scores in Familial Alzheimer Diseases
» #1 Local Synthesis of Dynein Cofactors Matches Retrograde Transport to Acutely Changing Demands
» #3 Relation of Dysglycemia to Structural Brain Changes in a Multiethnic Elderly Cohort
» First Place: White Matter Changes in Alzheimer's Disease
» Alzheimer's Association International Conference (AAIC 2016)
» #1 Neuronal activity enhances tau propagation and tau pathology
» #2 Sleep Disordered Breathing and White Matter Hyperintensities in Community-Dwelling Elders
» #2 Dementia Risk and Protective Factors Differ in the Context of Memory Trajectory Groups
» #2 Parkinson's Disease: Guilt by Genetic Association
» #1 PDE5 Exists in Human Neurons and is a Viable Therapeutic Target for Neurologic Disease
» #1 Extracellular Tau Oligomers Produce An Immediate Impairment of LTP and Memory
» #3 Mediterranean Diet and Brain Structure in a Multiethnic Elderly Cohort
» #2 Novel Selective Calpain 1 Inhibitors as Potential Therapeutics in Alzheimer's Disease
» #1 First Place: DREADDs Activation in the Medial Entorhinal Cortex (MEC) of EC-Tau Mice
» #1 F-box/LRR-repeat protein 7 is genetically associated with Alzheimer's disease
» #2 The keystone of Alzheimer pathogenesis might be sought in Aβ physiology
» #1 Stereotaxic Infusion of Oligomeric Amyloid-beta into the Mouse Hippocampus
» #1 SUMO1 Affects Synaptic Function, Spine Density and Memory
» #2 Connectivity and Circuitry in a Dish Versus in a Brain
» #2 Mediterranean Diet and Leukocyte Telomere Length in a Multi-ethnic Elderly Population
» Neurotherapeutics: Rethinking Alzheimer's Disease Therapies
» #2 Olfactory deficits predict cognitive decline and Alzheimer dementia in an urban community
» Regulation of synaptic plasticity and cognition by SUMO in normal physiology and Alzheimer's disease
» Lobar Microbleeds Are Associated with a Decline in Executive Functioning in Older Adults
» Targeting Axonal Protein Synthesis in Neuroregeneration and Degeneration
» Coding mutations in SORL1 and Alzheimer's disease
» Axonally Synthesized ATF4 Transmits a Neurodegenerative Signal across Brain Regions
» Neurological disorders: Quality-control pathway unlocked
» Estrogen Receptor β Variants Modify Risk for Alzheimer's Disease in a Multiethnic Female Cohort
» Local synthesis of TC10 is required for membrane expansion during axon outgrowth
» Dynamin 1 is Required for Memory Formation
» Biobanked Alzheimer's Brain Tissue Yields Living Neurons
» Picomolar Amyloid-ß Peptides Enhance Spontaneous Astrocyte Calcium Transients
 |  |  | ||
| Ronald K. H. Liem, PhD | Jian Zhao, PhD | Kristy R. Brown |
Charcot-Marie-Tooth disease (CMT) is an inherited peripheral neuropathy that affects 1:2500 people worldwide. Patients suffer from degeneration of the peripheral nerves that control sensory information of the foot/leg and hand/arm. Although over 90 causative genes have been identified, most do not yet have good animal models to investigate mechanisms of disease onset and progression. The scarcity of clinically relevant animal models is also hampering the development of treatments for the disease. The laboratory of Taub faculty member Dr. Ronald Liem has focused on CMT2E, which is caused by mutations in the neurofilament light gene, NEFL. A number of different dominant mutations in this gene can cause CMT2E. The Liem laboratory has made a knock-in mouse model containing the N98S mutation in Nefl. These NeflN98S/+ mutant mice reproduce many of the early-onset symptoms that have been described in patients with the N98S mutation, including problems in balance, tremor, cerebellar abnormalities, hearing defects and accumulations of neurofilamentous inclusions in spinal cord neurons. Most of these symptoms can already be observed in two week old mice. Studying this animal model is, therefore, of key importance to understand the mechanism and identify therapeutic targets.
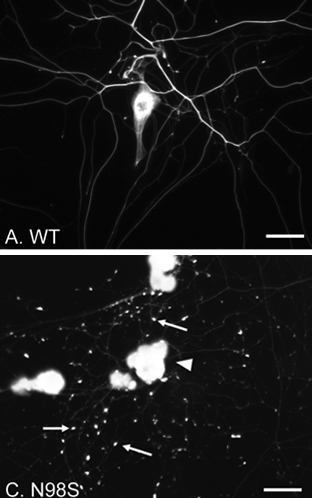
Figure 1. Immunofluorescence micrographs of cultured DRG neurons from Nefl+/+ and NeflN98S/+ mice labeling NFL.
(A and C) Low power views of DRG neurons from Nefl+/+ (A) and NeflN98S/+ (C) mice. Note that in NeflN98S/+ DRG neurons, the processes are characterized by large amounts of enlarged and bright particles along the processes, pointed by arrows (C). Scale bars = 50 μm
A study by Zhao et al. recently published in PLOS One conducted ultrastructural and immunofluorescence studies of dorsal root ganglia (DRG) from NeflN98S/+ mutant mice. Neurofilamentous inclusions were readily observed in cultures of adult DRG neurons stained with an antibody to the neurofilament light protein (Fig.1). Similarly, abnormal neurofilamentous inclusions were described in embryonic day 15 DRG cultures from the mutant animals. Electron microscopic analysis showed that these inclusions are made up of disordered neurofilaments packed in high density, segregated from other organelles. Immunochemical studies show decreased NFL protein levels in DRG, cerebellum and spinal cord in NeflN98S/+ mice, and total NFL protein pool is shifted toward the triton-insoluble fraction. These inclusions are believed to play an important role in affecting normal trafficking and function in peripheral neurons. This EM study reveals the nature of the inclusions in NeflN98S/+ mice. These results provide useful information to understand mechanisms of CMT2E disease, and identify DRG from NeflN98S/+ mice as a useful cell line model for therapeutic testing.
In summary, this study reveals the nature of inclusions in NeflN98S/+ mutant mice, and provides useful information in explaining how mutations in a cytoskeleton protein lead to neurodegeneration. More importantly, these studies establish a potential therapeutic approach to identify effective compounds to treat CMT diseases.
Ronald K. H. Liem, PhD
Professor of Pathology and Cell Biology (in the Taub Institute)
rkl2@cumc.columbia.edu
Jian Zhao, PhD
Associate Research Scientist, Department of Pathology and Cell Biology
jz2554@cumc.columbia.edu
Kristy R. Brown
Senior Staff Associate
krb1@cumc.columbia.edu
LTP and Memory Impairment Caused by Extracellular Aβ and Tau Oligomers is APP-Dependent

Ottavio Arancio, MD, PhD
The amyloid cascade hypothesis has dominated research on the origin of Alzheimer's disease (AD) over the last 20 years. According to the hypothesis, the protein beta-amyloid starts the disease by triggering pathology of another protein called tau, which then spreads the disease throughout the brain independently of beta-amyloid. The two proteins would, therefore, be placed in series, and this has been blamed for the failure of the many clinical trials attempting to cure the disease by interfering with beta-amyloid without blocking the negative effects of the downstream tau. This view is challenged in a recent work by Taub faculty member Dr. Ottavio Arancio, leading an international team of researchers, including the laboratories of Daniela Puzzo and Claudio Grassi at the University of Catania and the Catholic University of Rome in Italy, and the laboratories of Paul Fraser at the University of Toronto in Canada and Luciano D'Adamio at Albert Einstein in New York.
Starting from the discovery that a brief exposure to small soluble forms of beta-amyloid and tau, alone or in combination, dramatically disrupts memory and cell to cell communication, the team has found that beta-amyloid and tau, but not other proteins involved in other neurodegenerative diseases, require the presence of amyloid precursor protein (APP) to disrupt memory and cell to cell communication. Intriguingly, they also found that APP is necessary for the entrance of the two proteins within cells. Indeed, suppression of APP rescued the deleterious effects of both soluble beta-amyloid and tau on memory and cell to cell communication.
Thus, after this manuscript, the story about the origin of this devastating disorder needs to be told in a different manner. Beta-amyloid and tau are no longer placed in series as the amyloid cascade posits; instead, they should be placed in parallel in a series of negative events that could be initiated by the two proteins separately or in combination. Intriguingly, APP is at a key central point in the disease origin. The consequences of such a new sequence of events is that it is unlikely that therapies aimed at blocking the negative effects of beta-amyloid and tau alone will work. Most importantly, the research from the Arancio laboratory indicates that the field should look at APP as a target for therapies against Alzheimer's disease.
Ottavio Arancio, MD, PhD
Professor of Pathology and Cell Biology (in the Taub Institute)
oa1@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.