
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspectives:
March 2016
» #1 PDE5 Exists in Human Neurons and is a Viable Therapeutic Target for Neurologic Disease
» #2 PP2A Methylation Controls Sensitivity and Resistance to β-Amyloid–Induced Cognitive and Electrophysiological Impairments
» #2 PP2A Methylation Controls Sensitivity and Resistance to β-Amyloid–Induced Cognitive and Electrophysiological Impairments
PDE5 Exists in Human Neurons and is a Viable Therapeutic Target for Neurologic Disease
 |  | |
| Andrew Teich, MD, PhD | Ottavio Arancio, MD, PhD |
Phosphodiesterase 5 (PDE5) is a critical component of the cGMP-PKG axis of cellular signaling in neurons, and inhibition of PDE5 has been shown to be therapeutic in a wide range of neurologic conditions in animal models. Despite the promising results of PDE5 inhibitors as memory enhancers in models of central nervous system (CNS) diseases, there has been no major effort to translate these findings into the clinic. The reason is linked to skepticism by industry scientists towards the presence of PDE5 in adult human brain. This belief is based on only a few papers where PDE5 mRNA levels were found to be low or non-existent in adult human brain. No group has attempted to look for PDE5 protein in human brain, and as a result, many industry scientists are deeply skeptical of PDE5’s role in human neurophysiology.
In the present study, the Teich laboratory and the laboratory of Dr. Ottavio Arancio worked in collaboration to conclusively show that PDE5 is expressed in adult human brain, and that PDE5 is expressed in neurons. As recently published in the Journal of Alzheimer's Disease, they first show that past attempts to quantify PDE5 mRNA were flawed due to the use of incorrect primers, and that when correct primers are used, PDE5 mRNA is detectible in human brain tissue. Then, the investigators show that PDE5 protein exists in human brain by Western blot and ELISA. Most importantly, they perform immunohistochemistry, and demonstrate that PDE5 is present in human neurons. This was done to counter the frequent criticism that the little PDE5 that does exist in the brain is probably only found in the vasculature.
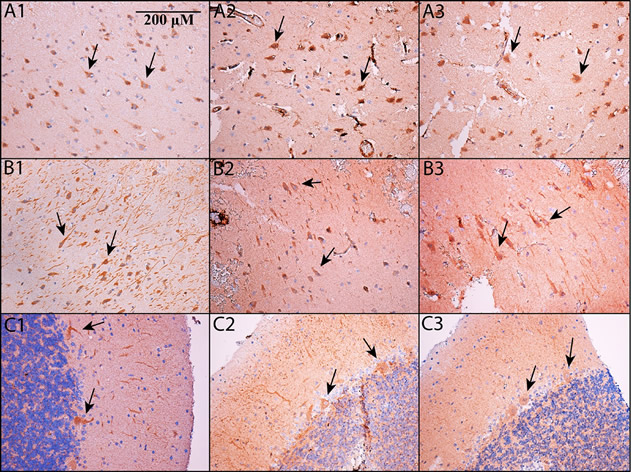 Fig. 2. PDE5 is expressed in neurons. Immunohistochemistry was performed on formalin-fixed, paraffin embedded sections of cortex (A1-A3), hippocampus (B1-B3; shown is subfield CA2/3 – see main text for details) and cerebellum (C1-C3). All images are shown at 200x magnification; the scale bar in Panel A1 applies to all panels. Panels A1, B1, and C1 use an Abcam antibody, panels A2, B2, and C2 use a Santa Cruz antibody, and panels A3, B3, and C3 use an Atlas antibody (see Methods). In all cases, PDE5 is expressed in the cytoplasm of neurons. In cortex and hippocampus, PDE5 is expressed primarily in large, pyramidal-type neurons (see arrows), whereas in cerebellum, it is prominent in Purkinje neurons (see arrows).
|
The work in this paper is uniquely important from a medical perspective. First of all, this work validates the relevance of a large body of animal research on PDE5 for human neurologic disease. Secondly, PDE5 inhibitors are widely used for other non-neurologic conditions, and so the side-effect profile of this class of drugs is mild and well characterized. Thus, PDE5 inhibitors have therapeutic potential for a variety of neurologic diseases, and the work presented here will stimulate future research in this field.
Andrew Teich, MD, PhD
Assistant Professor of Pathology and Cell Biology (in the Taub Institute)
aft25@cumc.columbia.edu
Ottavio Arancio, MD, PhD
Associate Professor of Pathology and Cell Biology (in the Taub Institute)
oa1@cumc.columbia.edu
 | | |
| Russell Nicholls, PhD | Ottavio Arancio, MD, PhD |
Beta-amyloid is thought to play a central role in the molecular etiology of Alzheimer's disease. However, despite considerable evidence to support this contention, we have only a poor understanding of the mechanisms by which beta-amyloid exerts its pathological effects. To address this gap and identify new potential therapeutic targets for Alzheimer's disease treatment or prevention, Taub faculty member Dr. Russell Nicholls conducted a series of experiments, published recently in PNAS, to examine the role of the enzyme protein phosphatase 2A (PP2A) in this process.
In this study, Dr. Nicholls and colleagues found that PP2A activity regulates the sensitivity of mice to the pathological actions of beta-amyloid. To demonstrate this, Dr. Nicholls collaborated with Nobel Laureate, Dr. Eric Kandel to generate two novel lines of transgenic mice that alter PP2A activity in vivo by targeting the methylation of its catalytic subunit. He then collaborated with members of the Arancio laboratory, as well as researchers at the University of Newcastle and the Baylor Research Institute, to examine the biochemical consequences of these manipulations and their effect on cognitive and electrophysiological impairments caused by beta-amyloid.
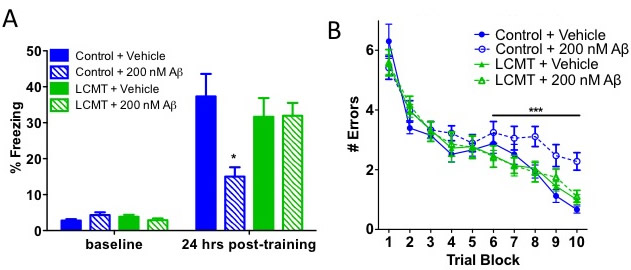 Fig. 4 A&B. LCMT-1 overexpression reduces behavioral and electrophysiological impairments caused by oligomeric Aβ. (A) Average percent of time spent freezing (± SEM) during initial exposure to the training context (baseline) and 24 h after foot shock for the indicated genotype and treatment groups Two-way ANOVA effect of training: F(1,43) = 134.51, P < 0.0001; training × group interaction: F(3,43) = 5.50, P < 0.01. Bonferroni post hoc comparisons of freezing at 24 h: P < 0.01 for Aβ- treated vs. vehicle-treated controls and P > 0.05 for LCMT + AB vs. vehicle- treated controls. P > 0.05 for all baseline freezing response comparisons (n = 11 or 12 animals per group). (B) Average number of errors committed (± SEM) during each three-trial training block of a 2-d radial arm water maze task for the in- dicated genotype and treatment groups (n = 11 or 12 animals per group). Two- way repeated-measures ANOVA with block and treatment as factors for control + vehicle vs. control + Aβ on day 2 (blocks 6–10): F(1,21) = 27.74; P < 0.0001 for treatment; P > 0.05 for control + vehicle vs. LCMT + Aβ and control vehicle vs. LCMT vehicle.
|
Their work showed that mice that over express the PP2A methylesterase, PME-1, were more sensitive to beta-amyloid induced impairments while mice that over express the PP2A methyltransferase, LCMT-1, were protected from these impairments. Neither genetic manipulation affected endogenous beta-amyloid levels, suggesting that PP2A methylation regulates beta-amyloid sensitivity by altering the response to beta-amyloid rather than beta-amyloid production. Moreover, these manipulations appeared to selectively alter the response to pathological concentrations of beta-amyloid since they did not affect baseline behavioral or electrophysiological responses, or the electrophysiological response to beta-amyloid when applied at physiological concentrations. Together, these data identify PP2A methylation as a key regulator of beta-amyloid sensitivity with unique potential as a novel therapeutic target for Alzheimer's disease prevention or treatment.
Russell Nicholls, PhD
Assistant Professor of Pathology and Cell Biology (in the Taub Institute)
rn95@cumc.columbia.edu
Ottavio Arancio, MD, PhD
Associate Professor of Pathology and Cell Biology (in the Taub Institute)
oa1@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.