
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspectives:
September 2015
» #1 F-box/LRR-repeat protein 7 is genetically associated with Alzheimer's disease.
» #2 The keystone of Alzheimer pathogenesis might be sought in Aβ physiology.
» #2 The keystone of Alzheimer pathogenesis might be sought in Aβ physiology.
View Archive
» #1 Stereotaxic Infusion of Oligomeric Amyloid-beta into the Mouse Hippocampus.
» #1 SUMO1 Affects Synaptic Function, Spine Density and Memory
» #2 Connectivity and Circuitry in a Dish Versus in a Brain
» #2 Mediterranean Diet and Leukocyte Telomere Length in a Multi-ethnic
Elderly Population
» Neurotherapeutics: Rethinking Alzheimer's Disease Therapies
» #2 Olfactory deficits predict cognitive decline and Alzheimer dementia in an urban
community
» Regulation of synaptic plasticity and cognition by SUMO in normal physiology and
Alzheimer's disease
» Lobar Microbleeds Are Associated with a Decline in Executive Functioning in
Older Adults
» Targeting Axonal Protein Synthesis in Neuroregeneration and Degeneration
» Coding mutations in SORL1 and Alzheimer's disease
» Axonally Synthesized ATF4 Transmits a Neurodegenerative Signal across Brain Regions
» Neurological disorders: Quality-control pathway unlocked
» Estrogen Receptor Œ± Variants Modify Risk for Alzheimer's Disease in a Multiethnic
Female Cohort
» Local synthesis of TC10 is required for membrane expansion during axon outgrowth
» Biobanked Alzheimer's Brain Tissue Yields Living Neurons
» Biobanked Alzheimer's Brain Tissue Yields Living Neurons
» Picomolar Amyloid-Œ≤ Peptides Enhance Spontaneous Astrocyte Calcium Transients
F-box/LRR-repeat protein 7 is genetically associated with Alzheimer's disease.
 |  |
| Giuseppe Tosto, MD, PhD | Richard Mayeux, MD, MSc |
Late-onset Alzheimer's disease (LOAD) is the leading cause of dementia in the elderly. The most common genetic risk factor is the APOE-ɛ4 allele, with an attributable risk of 10–15%. Over the past few years, several large consortium have identified up to 20 additional novel loci associated with increased risk of LOAD. However, aside from APOE, the effect sizes have been small, suggesting that a large part of the genetic component of LOAD still remains unexplained. Admixed populations like African Americans and Caribbean Hispanics have a unique genetic profile and higher incidence of the disease, which may facilitate the discovery of new genes and pathways in complex diseases such as LOAD.
In fact, previous work by Taub colleagues Drs. Richard Mayeux and Christiane Reitz identified the ABCA7 gene as a susceptibility locus with an effect size similar to that of APOE, in a genome wide association study (GWAS) of African Americans. Hispanics are one of the fastest growing minority groups in the U.S. with Caribbean Hispanics making up to 30% of this population. Taub researchers and other groups have demonstrated that known genetic and non-genetic risk factors have different effects on LOAD risk in this population, as compared to other ethnic groups.
In a study recently published in the Annals of Clinical and Translational Neurology, Drs. Giuseppe Tosto, Richard Mayeux, and colleagues from the Taub Institute examined potential novel genetic loci associated with LOAD in a large cohort of Caribbean Hispanics derived from three studies based at Columbia University: the Washington Heights-Inwood Columbia Aging Project (WHICAP); Estudio Familiar de Influencia Genetica en Alzheimer (EFIGA); and the Northern Manhattan Study (NOMAS). A novel locus on chromosome 5, single nucleotide polymorphism rs75002042 in the FBXL7 gene, was found to be significantly associated with LOAD in the case-control cohort (n= 4,514 individuals; OR=0.61, p-value=6.19E-09), and was further confirmed in an expanded cohort that included related family members (n=5300 individuals). Other variants linked to rs75002042 were found to be significant in two independent datasets of different ethnicity used for replication. Experiments performed in two models of AD-like transgenic mice found Fbxl7 protein overexpressed when compared to wild-type littermates. This finding was consistent with publicly available microarray studies: significant overexpression of Fbxl7 was confirmed in human LOAD brains compared to non-demented control brains.
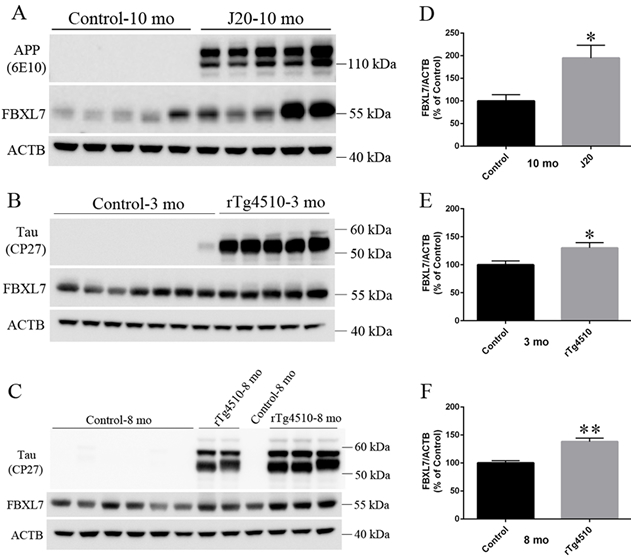 Figure 2. The expression of Fbxl7 protein in J20 and rTg4510 mice. Protein samples from the cortex and hippocampus of: (A) 10-months-old J20 (n = 5) and control mice (n = 5), (B) 3-month-old rTg4510 (n = 5) and control mice (n = 7), and (C) 8-month-old rTg4510 (n = 2) and control mice (n = 4) were separated in 4–12% Bis-Tris polyacrylamide gels and blotted with mouse primary antibodies against APP/Ab (6E10), Tau (CP27), Fbxl7, or ACTB. The quantitation of integrated density of Fbxl7 and ACTB in (A–C) was shown in (D–F), respectively. Data are presented as mean standard error of Fbxl7/ACTB. *P < 0.05
|
In addition to the new discovery of FBXL7, the current study was also able to confirm six known LOAD-associated susceptibility genes: ABCA7, CD33, CELF1, FERMT2, FRMD4A, and SLC24A4-RIN3. This further supports the role of these loci in this complex disorder of aging. Additional confirmation (e.g., sequencing and functional analyses) will be mandatory to validate and fully understand the role of FBXL7 in the pathogenesis of LOAD.
Giuseppe Tosto, MD, PhD
Postdoctoral Research Scientist (in the Taub Institute and the Gertrude H. Sergievsky Center)
gt2260@cumc.columbia.edu
Richard Mayeux, MD, MSc
Gertrude H. Sergievsky Professor of Neurology, Psychiatry and Epidemiology (in the Gertrude H. Sergievsky Center and in the Taub Institute)
rpm2@cumc.columbia.edu
The keystone of Alzheimer pathogenesis might be sought in Aβ physiology.

Ottavio Arancio, MD, PhD
The laboratory of Dr. Ottavio Arancio studies the cellular and molecular mechanisms of synaptic plasticity that underlie long-lasting changes in memory function, in both normal brains and in the brains of those affected by Alzheimer's disease (AD) and other neurodegenerative disorders. Over the course of his career, Dr. Arancio has remained particularly committed to pursuing original, basic and translational research studies that contribute new biological insights into the etiopathogenesis of AD and related disorders. Through both original research contributions and insightful, academic reviews, Dr. Arancio continues to challenge traditional assumptions about two long-debated proteins: amyloid-beta peptide (Aβ) for amyloid plaques and tau for neurofibrillary tangles, which have so dominated the field.
Recently published in the journal Neuroscience, Dr. Arancio and colleagues, including first author Daniela Puzzo (a former postdoc in the Arancio Laboratory), present another knowledgeable, penetrating review concerning over reliance on Aβ pathogenesis and the amyloid cascade hypothesis, in particular. Why does a protein that is physiologically produced in the healthy brain, at some point, increase, and why do certain individuals present an increase of Aβ levels or plaque deposits without any sign of clinical dementia? Dr. Arancio and co-authors propose that a deeper understanding of AB physiology might shed some much needed insight on these enduring questions. They suggest that, in physiological conditions, synaptic activity triggers Aβ release which, in turn, modulates receptors for nicotine that are located at the synapse leading to an enhancement of neurotransmitter release, with a consequent increase of synaptic plasticity and memory. Conversely, they speculate that when Aβ cannot exert its physiological functions (i.e., for a receptor resistance) a negative feedback mechanism would induce a compensatory increase of its production leading to an abnormal accumulation that reduces nicotinic receptor function, leading to synaptic dysfunction and memory loss. According to the authors, "overtime, if not brought back to its normal homeostasis, this chronic failure would produce a reduction of the protein levels due to 'cellular exhaustion'."
In addition, the authors argue that indiscriminate Aβ removal would interfere with neuronal homeostasis, further impoverishing learning and memory. In consideration of the fact that clinical trials based on lowering Aβ levels have proven largely unsuccessful, and in advance of the continued pursuit of new therapeutic strategies based on the removal of Aβ from the brain, the authors conclude with an imperative that "a different vision is needed to build the bridge between [the] physiological and pathological role of Aβ." Read More from Puzzo et al.
Ottavio Arancio, MD, PhD
Associate Professor of Pathology and Cell Biology (in the Taub Institute)
Oa1@columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.