
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspectives:
December 2014
» Regulation of synaptic plasticity and cognition by SUMO in normal physiology and Alzheimer's disease
» Lobar Microbleeds Are Associated with a Decline in Executive Functioning in Older Adults
» Lobar Microbleeds Are Associated with a Decline in Executive Functioning in Older Adults
Regulation of Synaptic Plasticity and Cognition by SUMO in Normal Physiology and Alzheimer's Disease

Ottavio Arancio, MD, PhD
Post-translational modifications constitute an important step in protein biosynthesis that regulates protein function by attaching functional groups to the amino-acids that compose the protein. A new study from the laboratory of Taub investigator Octavio Arancio, PhD, published this month in Scientific Reports, demonstrates that attachment of the Small Ubiquitin-like MOdifier (SUMO) group to proteins is a post-translational modification required for normal long-term synaptic plasticity, the underlying cellular correlate for learning and memory, as well as cognitive processes. Furthermore, Lee et al shows that this regulatory mechanism is disrupted in Alzheimer's disease (AD), which implicates SUMOylation in both normal neural physiology and disease pathogenesis. The demonstration that SUMO has such a broad role in synaptic functioning places SUMOylation among central regulatory mechanisms such as phosphorylation. As such, these results are likely to have a large impact on the neuroscience field.
The Arancio laboratory has discovered that SUMOylation is reduced in brain tissue from AD patients. While neuronal activation normally induces up-regulation of SUMOylation, this effect is impaired by amyloid-beta, a toxic protein that is known to play a major role in AD. Amyloid-beta is also a known potent disruptor of synaptic function. However, enhancing SUMOylation via transduction of its conjugating enzyme, Ubc9 (see figure), rescues amyloid-beta-induced deficits in synaptic plasticity and memory. These novel findings strongly implicate impaired SUMOylation as a central pathway in Alzheimer's disease pathogenesis and present an exciting new avenue for potential drug discovery.
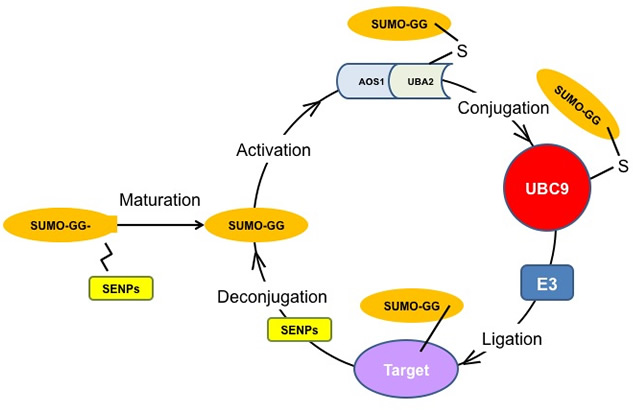 FIGURE LEGEND: The SUMOylation Pathway. The SUMO enzymatic cascade catalyzes the dynamic post-translational modification process of transfer of SUMO protein to other proteins (SUMOylation). The Small Ubiquitin-related Modifier, SUMO, is conjugated to its substrates through three sequential enzymatic steps: activation, involving the E1 enzyme, AOS1/UBA2; conjugation, involving the E2 enzyme, UBC9; substrate modification, through the cooperation of the protein ligases, E2 and E3. De-conjugation (De-SUMOylation) occurs through enzymes called SENPs, which recycle free SUMO and unmodified target proteins. Lee et al have demonstrated that the pathway is down-regulated in Alzheimer's disease. This work gives the impetus to the development of drugs acting at different steps of the pathway to alleviate AD symptomatology. |
When Lee et al first started this project, very little was known about the role of SUMO in the brain, since this protein modifier is relatively newly-discovered in comparison to others. Major questions were unanswered, including whether SUMOylation was involved in the signaling networks that underlie learning and memory. The Arancio laboratory has now addressed this prominent question and, additionally, extended the discovery with translational relevance for Alzheimer's disease.
Ottavio Arancio, MD, PhD
Oa1@columbia.edu
Lobar Microbleeds Are Associated with a Decline in Executive Functioning in Older Adults
 |
 |
| Adam M. Brickman, PhD | Irene B. Meier |
Over the past several decades, there have been myriad efforts to characterize the nature of age-associated cognitive decline. Most studies agree that aging is associated with a gradual loss of cognitive abilities, with increasing variability or individual differences. Aging, per se, is not a cause of cognitive decline but rather a variable that likely captures multiple accumulating biological changes over time that collectively affect our mental abilities. There are currently no accepted definitions of what constitutes "normal" versus "pathological" cognitive aging. Typically, we consider normal cognitive aging as decline in certain cognitive abilities in the absence of frank disease or evidence of neurodegenerative pathology. Several authors have argued that small vessel cerebrovascular disease is a primary source of normal cognitive aging. "Pathological cognitive aging," on the other hand, typically refers to cognitive decline that is due to a neurodegenerative condition. In the case of Alzheimer's disease (AD), the most common cause of pathological aging, current hypothetical pathogenic models emphasize the precipitating role of beta amyloid protein and tau pathology, which ultimately cause neurodegeneration and the neuropsychological syndrome associated with the disease. The amyloid cascade hypothesis suggests that imbalance of production and clearance of Aβ is a primary driver of pathological cognitive aging.
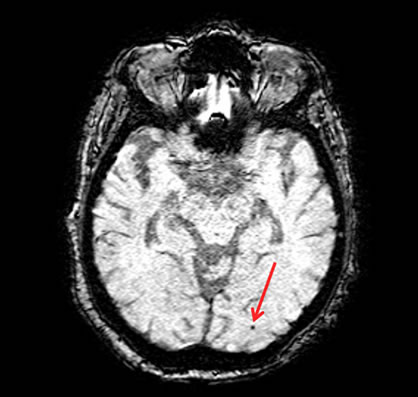
Figure 1 is an example of a lobar microbleed on a gradient echo (GRE) MRI scan.
Recent literature, however, implicates small vessel cerebrovascular disease as an important source of dysfunction in AD and perhaps disease pathogenesis, suggesting some overlap between factors that promote cognitive decline due to normal aging and due to AD. Cerebral amyloid angiopathy, or the deposition of beta amyloid in blood vessel walls, may represent a mechanistic link between amyloid-centered hypothesis and the increasing attention towards vascular factors in AD pathogenesis – promoting cognitive decline in individuals without dementia but also contributing to the pathogenesis of AD. Cerebral amyloid angiopathy manifests on MRI scans as cerebral microbleeds distributed in lobar regions. Thus, lobar microbleeds are considered a radiological indicator of the presence of underlying cerebral amyloid angiopathy and associations with clinical characteristics involving microbleeds can be attributed to the role of the underlying amyloid pathology. Over the past several years, we have been examining the intersection between markers of cerebrovascular pathology and Alzheimer's disease. In the current study, published this month in the journal Cerebrovascular Diseases, we used a retrospective longitudinal design to examine whether the presence of lobar microbleeds, as a marker of cerebral amyloid angiopathy, is associated with cognitive decline.
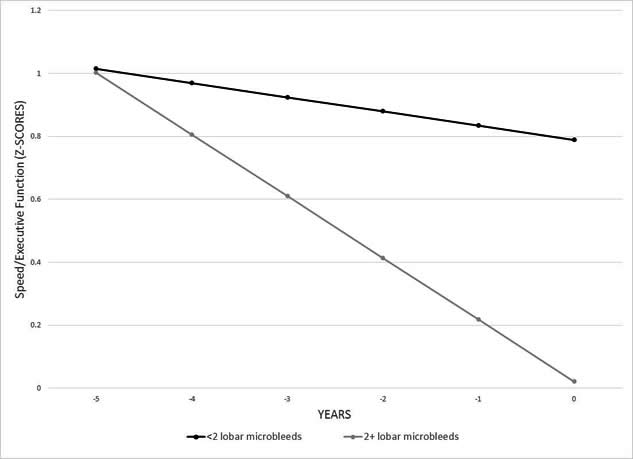 Figure 2 shows great amounts of executive functioning decline in individuals with (2+) vs. without microbleeds. |
Participants came from an ongoing longitudinal community-based aging study, in which subjects are evaluated at 18-24 month intervals and received a full medical, neurological, and neuropsychological examination at each of the follow-up visits. MRI scans were available on 197 non-demented participants. Microbleeds were rated visually and cognition was assessed with a neuropsychological battery, providing summary scores for memory, language, executive, and visuospatial abilities. We examined longitudinal change in cognition beginning about 9 years before the MRI scan in people with 2 or more and fewer than 2 lobar microbleeds. We found that individuals with 2 or more lobar microbleeds had worse executive functioning at the visit closest to the MRI scan and had faster decline in executive functioning over time than individuals with fewer than 2 microbleeds.
Our findings show that lobar microbleeds, a marker of cerebral amyloid angiopathy, are associated with an accelerated rate of executive function decline. The presence of cerebral amyloid angiopathy may be an important source of cognitive decline in aging and an important link between normal cognitive aging and Alzheimer's disease.
Adam Brickman, PhD
amb2139@cumc.columbia.edu
Irene Meier
Ibm2106@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.