
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspectives:
April 2016
» #1 White Matter Hyperintensities are a Core Feature of Alzheimer's Disease: Evidence From the Dominantly Inherited Alzheimer Network
» #2 Parkinson's Disease: Guilt by Genetic Association
» #2 Parkinson's Disease: Guilt by Genetic Association
 |  | |
| Adam M. Brickman, PhD | Seonjoo Lee, PhD |
White matter hyperintensities (WMH) are areas of increased signal visualized on certain types of magnetic resonance imaging (MRI) scans of the brain. They are thought to reflect small vessel cerebrovascular disease. Previous work from Dr. Brickman's laboratory and others has shown that the severity of WMH is associated with risk, onset, and progression of Alzheimer's disease (AD). These observations are often interpreted as evidence that small vessel cerebrovascular disease plays an additive role in clinical AD, contributing to the severity of symptomatology but not to the pathogenesis of the disease. However, there is considerable debate on the subject, with viable theoretical models that implicate a role of small vessel cerebrovascular abnormalities in AD pathogenesis, as well as emerging research suggesting interactions between WMH and primary AD pathology.
The study of the emergence of WMH, or any biological markers, and their contributions to AD in humans is difficult because the ordering and timing of the biological changes that lead to dementia can occur up to decades before the onset of symptoms. Furthermore, WMH are also tightly linked to vascular risk factors and age, so determination of their contribution to AD is potentially confounded by these factors. To overcome these issues, Dr. Brickman and colleagues turned to the landmark Dominantly Inherited Alzheimer Network (DIAN) study, which enrolls individuals at 50% risk for autosomal dominant AD by virtue of having a first-degree relative with a pathogenic mutation in one of three AD-causing genes (APP, PSEN1, or PSEN2). The purpose of the study was to examine the severity and distribution of WMH in presymptomatic mutation carriers to determine the extent to which WMH manifest in individuals genetically determined to develop AD. The study included 299 individuals (mean age=39-years-old) in whom 61.5% had a mutation that results in AD and 38.5% were first-degree relatives who were noncarrier controls. The estimated number of years to symptom onset was estimated by subtracting the affected parent's symptom onset age from the participant's age. Baseline MRI scans were analyzed for total and regional WMH and statistical models were constructed to examine WMH differences between mutation carriers and noncarriers with respect to the estimated number of years to symptom onset.
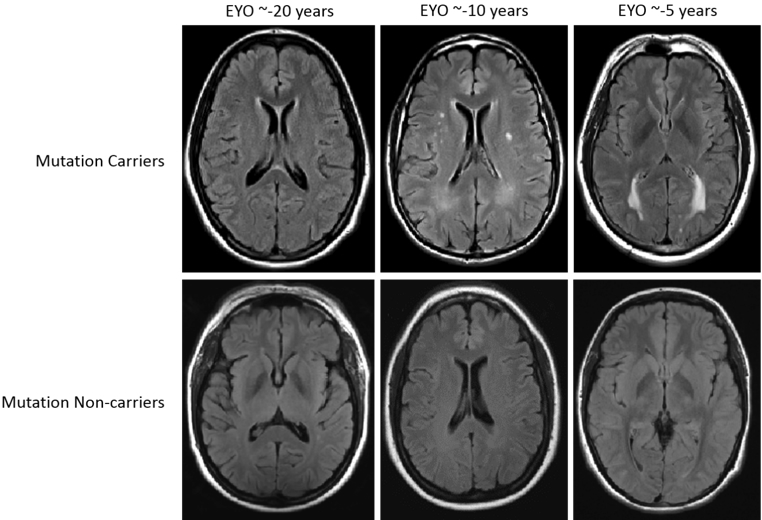 Figure 1. Examples of WMH distribution in mutation carriers (upper row) and non-carriers (lower row) across three EYO time points. The top row displays examples of T2-weighted FLAIR MRI scans from three mutation carriers at varying estimated years from symptom onset. The bottom row displays examples of MRI scans from non-carriers matched for estimated years from symptom onset (based on parental age of onset). All participants displayed in this figure had CDR scores of 0 at the time of MRI scan.
|
Recently published in the Annals of Neurology, results from the study showed that mutation carriers had greater total WMH volumes, which appeared to increase approximately 6 years prior to expected symptom onset. When examining the distribution of WMH regionally, the effects were most prominent in the parietal and occipital lobes, which showed divergent effects as early as 22 years before expected symptom onset. The findings demonstrate that autosomal-dominant AD is associated with increased WMH well before expected symptom onset, and suggest the possibility that small vessel cerebrovascular disease is a core feature of AD, a potential therapeutic target, and a factor that should be integrated in pathogenic models of the disease.
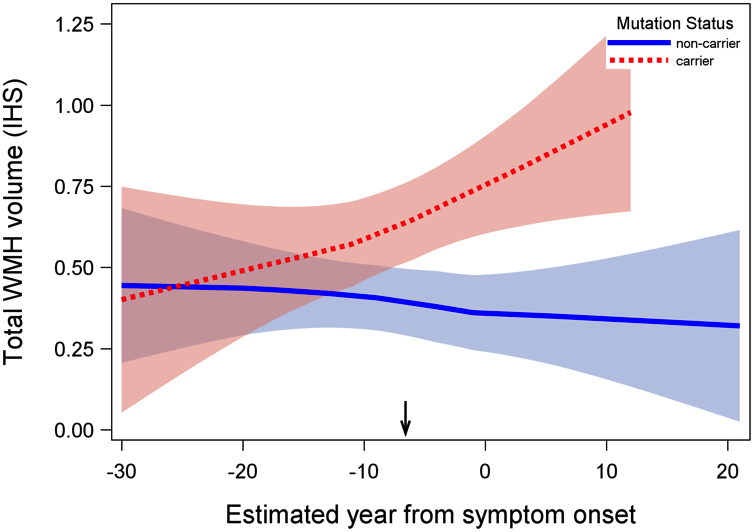 Figure 2. Association between estimated year from symptom onset and total WMH volume in mutation carriers and non-carriers. Mutation carriers had greater total WMH volume; differences in WMH volume between groups began increasing systematically approximately 6.6 years prior to estimated symptom onset (inflection point: -6.6 EYO, indicated by arrow on x-axis). Shaded areas represent 95% confidence intervals. Arrow indicates the inflection point in the analysis. IHS=inverse hyperbolic sine transformation.
|
Adam M. Brickman, PhD
Associate Professor of Neuropsychology (in Neurology, the Taub Institute, and the Gertrude H. Sergievsky Center)
amb2139@cumc.columbia.edu
Seonjoo Lee, PhD
Assistant Professor of Clinical Biostatistics (in Psychiatry)
sl3670@cumc.columbia.edu
Parkinson's Disease: Guilt by Genetic Association
 |  | |
| Asa Abeliovich, MD, PhD | Herve Rhinn, PhD |
In a brief review in Nature, Drs. Asa Abeliovich and Herve Rhinn discuss the utility of a sophisticated gene editing technology, termed CRISPR-Cas9, in the analysis of mechanisms of complex diseases such as Parkinson's disease (PD). Genome wide association studies from groups including Taub faculty members Drs. Lorraine Clark, Karen Marder, and many others have identified common variants (single nucleotide polymorphisms [SNPs], present in over ~5% of a population) across the human genome that are associated with disease risk, but just what these variants due has been difficult to parse. These studies present a number of challenges. For instance, swaths of the human genome are co-inherited across generations and typically include many common variants, and it is difficult to distinguish innocent bystanders that are co-inherited within such blocks of human genome from the truly causative genomic variants. Furthermore, the effects of such variants on human disease risk are typically pretty modest.
CRISPR-Cas9 technology in human embryonic stem (ES) cells offers a potential approach to very precisely modify the genome at a single variant and then determine the consequence. Abeliovich and Rhinn describe work from Rudy Jaenisch's lab which is focused on variants near the 3' end of the a-Synuclein gene. These are among the most strongly associated with PD across the human genome. aSynuclein protein is known to accumulate in PD brain, and the Jaenisch group focused on SNPs within the aSynuclein gene that have previously been shown to harbor epigenetic markings consistent with gene regulatory elements in human brain DNA. Using CRISPR-Cas9 technology, the Jaenisch group succeeded in converting human ES cells from the low-risk to the high-risk variant at a single suspected causative risk SNP, and showed that this led to a significant increase in gene expression. They, therefore, hypothesize that this SNP is part of the mechanism by which risk variants at the locus lead to an increase in disease risk. However, the work also clearly shows that other mechanisms must be at play, and this is an active area of research.
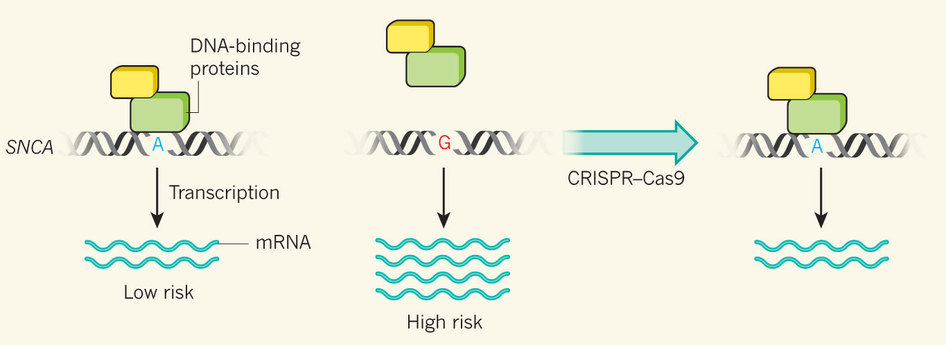 Figure. At one nucleotide in a non-protein-coding region of SNCA, the gene that encodes α-synuclein, the presence of the base adenine (A) is protective against Parkinson's disease, whereas the presence of another, guanine (G), confers increased risk. Soldner et al.3 report that this region regulates SNCA expression levels. If the two copies of the chromosome in a human cell each contain a different base at this site, gene expression is significantly higher from the risk-variant chromosome, owing in part to a reduction in the attachment of DNA-binding proteins that inhibit transcription. Using CRISPR–Cas9 gene-editing technology to remove the G and replace it with A reduces SNCA expression.
|
Asa Abeliovich, MD, PhD
Associate Professor of Pathology and Cell Biology, and Neurology (in the Taub Institute)
aa900@cumc.columbia.edu
Herve Rhinn, PhD
Assistant Professor of Pathology and Cell Biology
hr2239@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.