
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspectives:
January 2020
1: » Microglial Activation, but not Tau Pathology, is Independently Associated with Amyloid Positivity and Memory Impairment
2: » CRISPR/Cas9 Editing of APP C-Terminus Attenuates β-Cleavage and Promotes α-Cleavage
2: » CRISPR/Cas9 Editing of APP C-Terminus Attenuates β-Cleavage and Promotes α-Cleavage
 |
 |
| James Zou | William C. Kreisl, MD |
Microglia, the resident immune cell of the brain, has long been thought to be involved in the pathogenesis of Alzheimer’s Disease (AD). Microglia, depending on phenotype, can either have a protective role (phagocytosing amyloid plaques) or a harmful one (potentially propagating tau microfilaments or releasing proinflammatory molecules). It is currently not well understood which of these roles predominates in the development of AD. 11C-PBR28 is a second generation PET radioligand which can measure microglial activity in vivo, and has been found to correlate with severity and progression of disease. Recent in-vivo studies of neuroinflammation have shown somewhat conflicting results, suggesting that there may be both an early and late peak in microglial activation or that it increases through the disease process.
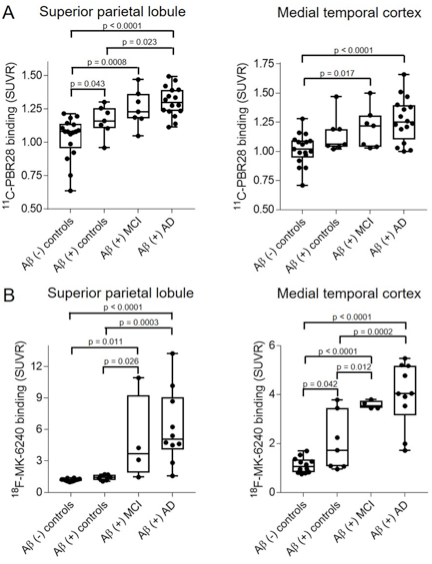
Figure 1: Pairwise comparison of SUVR values for subjects stratified across the AD clinical spectrum for both 11C-PBR28 (A) and 18F-MK6240 (B) in select regions. While microglial activation seemed to increase in a stepwise manner in neocortical regions but not medial temporal cortex, the opposite pattern was observed with tau deposition.
Recently published in Neurobiology of Aging, a new study by Dr. William Kreisl and colleagues from Neurology and Taub sought to determine how microglial activation was related to early (amyloid deposition) and late (memory impairment) signs of Alzheimer’s disease (AD). Led by first author James Zou, a senior VP&S medical student mentored under Dr. Kreisl via an extension of Neurology’s NIH T35 BRAIN program, the investigators performed amyloid PET and cognitive testing to determine amyloid and memory status, respectively. In 57 participants, 11C-PBR28 PET was acquired to measure microglial activation, and in 43 participants 18F-MK6240 PET was acquired to measure tau deposition. Using a multivariate analysis of variance (MANOVA), Zou et al. found that while both increased microglial activation and tau deposition were associated with amyloid deposition and cognitive impairment, only microglial activation was independently associated with both conditions. In addition, they found that while microglial activation seemed to increase in a stepwise manner in cortical regions only, this trend was found only in medial temporal regions for tau deposition.
These findings suggest that, in the absence of cognitive symptoms, amyloid deposition has a greater association with microglial activation than tau deposition, and that microglial activation has a role in both the early and late phases of AD pathogenesis. Further investigation into the role of microglia in AD pathogenesis may provide valuable insights into potential therapeutic interventions.
James Zou
4th Year Medical Student, Columbia University Vagelos College of Physicians and Surgeons
Jsz2108@cumc.columbia.edu
William C. Kreisl, MD
Boris and Rose Katz Assistant Professor of Neurology (in the Taub Institute)
wck2107@cumc.columbia.edu
CRISPR/Cas9 Editing of APP C-Terminus Attenuates β-Cleavage and Promotes α-Cleavage
 |  |  |
|||
| Amanda Snead | So Yeon Koo | Andrew A. Sproul, PhD |
CRISPR-mediated genome editing could potentially be used as a therapeutic tool to treat neurodegenerative diseases such as Alzheimer’s disease (AD). As a proof of principle, a study published in Nature Communications by senior author Dr. Subhojit Roy at the University of Wisconsin (now at UCSD) and collaborators, including members of Dr. Andrew Sproul’s laboratory in the Taub Institute, demonstrated that CRISPR-editing of the C-terminus of APP could reduce β-secretase cleavage of APP while promoting α-cleavage. This in turn leads to significant reduction in the production of toxic Aβ and β-CTF (C99) fragments from APP. Importantly, this strategy targeted the last coding exon of APP which spared the edited transcripts from destruction by nonsense-mediated decay (NMD) mechanisms and resulted in a truncated protein that in theory could still perform at least some physiological functions of APP. In addition, by targeting the distal C-terminus of APP rather than gene-correcting specific familial early-onset AD (EOAD) mutations, this approach could potentially be used more broadly for any EOAD autosomal-dominant mutation carrier.
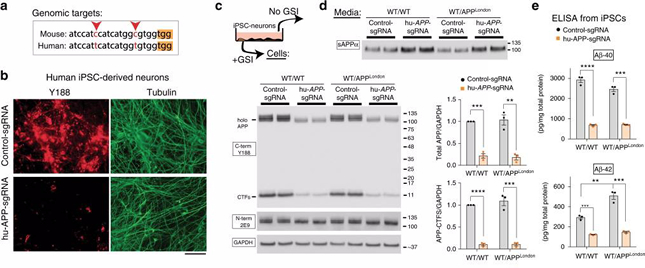 Figure: Gene editing of APP C-terminus and effects on APP processing in human cells. a Comparison of mouse and human APP-sgRNA targeting sequences (red arrowheads indicate differences; yellow bar denotes the PAM site). b Human iPSC-derived NPCs were transduced by lentiviral vectors carrying hu-APP-sgRNA and Cas9 (or non-targeting control-sgRNA/Cas9 as control) and differentiated into neurons. After 3 weeks of differentiation, cells were immunostained with the Y188 and Tuj1 (tubulin) antibodies. Note decreased APP (Y188) fluorescence, indicating APP editing. Scale bar 50 μm. c The iPSC-derived neurons above (or isogenic APPV717I London-mutant knock-in iPSC-neurons) were transduced and differentiated as above and immunoblotted with C- and N-terminus antibodies (GSI was added to allow detection of accumulated APP CTFs). Note attenuation of APP signal with Y188 after hu-APP-sgRNA treatment in both wild type and isogenic APPLondon iPSCs (quantified on right, mean ± SEM of three independent experiments). d Media from the iPSC-derived neurons above was immunoblotted for secreted sAPPα (6E10 antibody). Note increased sAPPα in sgRNA-treated samples, indicating upregulation of the non-amyloidogenic pathway. e ELISA of media from iPSC derived neurons. Note decreased Aβ in the sgRNA-treated samples (mean ± SEM of three independent experiments). Taken from Nature Communications volume 10, Article number: 53 (2019).
|
Dr. Roy’s group established this approach in cellular models as well as in vivo in mice. When his lab attempted to publish these findings, reviewers required human neuron data from an AD iPSC model. Thus, a collaboration began between the labs of Dr. Roy and Dr. Sproul to further validate these findings using an unpublished CRISPR-mediated knockin iPSC model of the APPLon (V717I) mutation generated by the Sproul lab. This work was conducted by former Sproul lab members Ms. Amanda Snead (currently at the University of Illinois Chicago in an MD/PhD program) and Ms. So Yeon Koo (currently in a PhD program at Weill Cornell). Neural progenitors (NPCs) derived from APPLon mutant and isogenic control iPSCs were treated with lentiviral particles containing either a control sgRNA or a sgRNA targeting APP (along with Cas9) and were subsequently differentiated into cortical neurons. Dr. Sproul’s group was able to successfully extend the initial findings to control and EOAD neurons. Dr. Roy’s lab is continuing to develop this approach as a potential therapeutic, while Dr. Sproul’s lab is using these tools to further elucidate mechanistic links between aberrant APP processing and tau dysregulation.
Andrew A. Sproul, PhD
Assistant Professor of Pathology and Cell Biology (in the Taub Institute) at the Columbia University Medical Center
Director of Stem Cell and Cellular Models Platform
aas2003@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.