
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
Featured Research
In the Lab:
Catherine Marquer, PhD

Catherine Marquer, PhD
A growing set of evidence from human genetic studies indicates that variants in genes involved in lipid metabolism and endolysosomal trafficking are risk factors to develop Alzheimer's disease. Maintaining the correct function of the endolysosomal pathway is a key feature of cellular homeostasis, as it regulates not only the uptake but also the correct targeting to recycling, degradation, or extracellular export of essential small molecules and proteins. This is even more critical in cells like neurons, with elongated and highly specialized subregions (i.e. dendrites vs. axon). In my lab, we focus on how lipids of the endolysosomal pathway can regulate its function, in both healthy and diseased states. These lipids include cholesterol, phosphoinositides, and BMP (also called LBPA). Our goal is to provide insight into Alzheimer's disease onset and progression, as well as possible therapeutic options.
My earlier post-doctoral in vitro and in vivo studies have led to the hypothesis that membrane cholesterol increase could be one of the earliest changes in late-onset Alzheimer's disease. Indeed, acutely increasing the membrane cholesterol burden of neurons in culture or inhibiting CYP46 activity and thus blocking brain cholesterol efflux in vivo recapitulates most Alzheimer's disease phenotypes, including increased toxic Aβ42 secretion, deficits in APP vesicular transport in neuronal processes, enlargement of early endosomes, tau hyper-phosphorylation, hippocampal atrophy, epileptic-like seizures, and cognitive deficits (Marquer et al., 2011; Marquer et al., 2014; Chali et al., 2015; Djelti et al., 2015). This work was performed with Dr. Potier, in collaboration with Drs. Cartier and Miles, at the ICM Institute, in Paris.
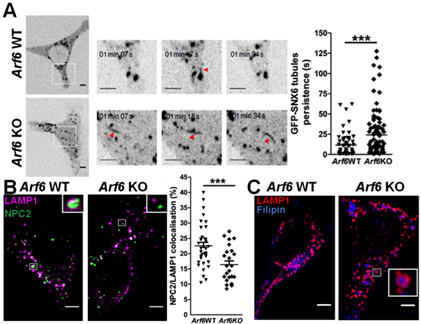
Figure 1. Cholesterol is redistributed to late endosomes/lysosomes in Arf6 KO fibroblasts as a result of aberrant retromer tubulation and NPC2 mistrafficking. (A) Left panel, spinning-disk confocal imaging of Arf6 WT and KO mouse embryonic fibroblasts (MEFs) expressing EGFP-SNX6. Scale bar=5 μm and 1 μm (insets). Arrows indicate tubules. Right panel, quantification of the persistence of EGFP-SNX6 tubules. *** stands for p<0.001 in t test with Welch's correction. (B) Representative immunostaining for LAMP1 (magenta), a late endosome-lysosome marker, and NPC2 (green) of Arf6 WT or KO MEFs. Scale bar=5 μm. Right panel, NPC2/LAMP1 colocalization quantification. *** stands for p<0.001 in Student’s t-test. (C) Representative immunostaining for LAMP1 (red) of Arf6 WT or KO MEFs labeled with filipin (blue), a fluorescent cholesterol ligand. Scale bar=5 μm.
A more recent study, performed when I was an associate research scientist in the Taub Institute with Dr. Gil Di Paolo, highlights how trafficking and lipid homeostasis can be intertwined. This work established a novel role for the small GTP-ase ARF6 in the control of a pathway mechanistically linking retromer function and cholesterol homeostasis (Marquer et al., 2016). This is particularly relevant for Alzheimer's disease, as retromer dysregulation is also an early alteration in its pathogenesis. The retromer complex plays an essential role in the trafficking of APP and its β- and γ-secretases. As Arf6 ablation is embryonically lethal in the mouse, we generated a new conditional Arf6 knockout (KO) model, the Arf6 flox/flox (fl/fl) mouse, and novel inducible Arf6 KO cellular models. We showed that Arf6 ablation resulted in a selective increase of an endosomal pool of phosphatidylinositol-4-phosphate (PI4P). This caused a perturbation of the tubulation of the retromer/SNXs complex, and the subsequent mistrafficking of its cargoes, including cation-independent mannose-6-phosphate receptor (CI-M6PR) and its ligand NPC2. Importantly, NPC2 mediates the transfer of cholesterol from the lumen of late endosomes/lysosomes (LE/Lys) to NPC1, a resident protein of the LE/Lys limiting membrane. The latter mediates the egress of cholesterol from the endolysosomal system, allowing for its distribution to other cellular compartments. Arf6 ablation resulted in NPC2 mislocalization away from LE/Lys and the subsequent accumulation of cholesterol in these compartments, as followed by staining with filipin, a fluorescent antibiotic that specifically binds to free cholesterol (see Figure 1).
Currently, the questions we are trying to address in my lab are:
-
1. Can we find new lipid-centric pathways and mechanisms to regulate endolysosomal function in neurons?
- 2. How do cellular lipid and endolysosomal modifications translate in terms of function or toxicity at the network/whole brain level in murine models and in humans?
- 3. What can we learn from lysosomal storage disorders to expand our understanding of Alzheimer's disease, and vice versa?
To answer these questions, we use a wide array of techniques, including quantitative confocal, spinning-disk and super-resolution microscopies, HPLC dosage of phosphoinositides and lipidomics (in collaboration with Dr. Estela Area Gomez). We also have a number of ongoing collaborations with members of the Taub Institute with complementary expertise, such as Drs. Joseph Lee and Sandra Barral (for human genetics), Dr. Abid Hussaini (for in vivo electrophysiology) and Dr. Andrew Sproul (for stem cell models). The Marquer lab is supported by funding from the NIH (NIA).
 From left to right, members of the Marquer lab include Dr. Catherine Marquer and Paola Piroli.
|
Catherine Marquer, PhD
Assistant Professor of Pathology and Cell Biology (in the Taub Institute)
cm3244@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.