
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
TaubCONNECT Research Perspectives:
October 2017
#1 Intra-Axonal Synthesis of SNAP25 is Required for the Formation of Presynaptic Terminals
#2 Increased Localization of APP-C99 in Mitochondria-associated ER Membranes Causes Mitochondrial Dysfunction in Alzheimer Disease
#2 Increased Localization of APP-C99 in Mitochondria-associated ER Membranes Causes Mitochondrial Dysfunction in Alzheimer Disease
Intra-Axonal Synthesis of SNAP25 is Required for the Formation of Presynaptic Terminals
 |  |  | ||
| Ulrich Hengst, PhD | Andreia F. R. Batista | José C. Martínez |
The formation of synapses is fundamental for the establishment of functional circuitry in the nervous system. Upon contact of axons with target-derived adhesive or soluble factors, presynaptic proteins cluster at the contact sites leading to the formation of presynaptic terminals. A new study by the laboratory of Ulrich Hengst, published in Cell Reports, sheds new light on a fundamental question in neurodevelopment: What is the source of the proteins from which the presynaptic terminals are built? In the current model, all proteins are transported from the cell body and rapidly recruited to sites of synaptic contact. First author Andreia Batista and José Martínez hypothesized that an alternative source for presynaptic proteins, intra-axonal synthesis, might be required as well.
The authors found that presynaptic organizing signals trigger local protein synthesis at their sites of contact with axons. The clustering of presynaptic proteins was prevented when protein synthesis inhibitors were applied specifically to axons. Previously, the transcript coding for the t-SNARE protein SNAP25 had been found in axonal transcriptome and translatome datasets. Here, the team of Dr. Hengst found that SNAP25 mRNA gets recruited to nascent presynaptic sites and locally translated. Axon-specific knockdown of SNAP25 mRNAs prevented the clustering of SNAP25 and other presynaptic proteins, and interfered with the release dynamics of synaptic vesicles. Together, these results establish that the induced intra-axonal synthesis of SNAP25 is a necessary, early step for the clustering of presynaptic proteins and the formation and function of presynapses.
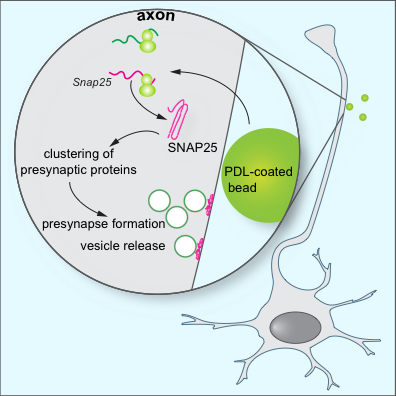
Figure 1: Model for the role of local SNAP25 synthesis in synapse formation. Batista et al. find that during the assembly of presynaptic terminals, mRNA translation is upregulated at the nascent presynapses. Local SNAP25 synthesis is required for the proper formation of presynaptic terminals and synaptic vesicle release.
This process is probably not only important during the formation of synapses but also in the mature nervous system because SNAP25 continues to be produced at already established synapses. These results will be important for the understanding of SNAP25 regulation and, more generally, protein homeostasis at synapses. Ongoing research in the laboratory of Dr. Hengst investigates the role of local SNAP25 synthesis at mature synapses, and how this process is affected under pathological conditions such as Alzheimer’s disease.
Ulrich Hengst, PhD
Associate Professor of Pathology and Cell Biology (in the Taub Institute for Research on Alzheimer's Disease and the Aging Brain
uh2112@cumc.columbia.edu
Andreia F. R. Batista
Graduate Student, MD/PhD program, Universidade do Minho, Portugal
andreiarodriguesbatista@gmail.com
José C. Martínez
Graduate Student, MD/PhD Program
jcm2216@cumc.columbia.edu
 |  | |
| Estela Area Gomez, PhD | Eric A. Schon, PhD |
In the amyloidogenic pathway associated with Alzheimer disease (AD), the amyloid precursor protein (APP) is cleaved by β-secretase to generate a 99-aa C-terminal fragment (C99) that is then cleaved by γ-secretase to generate the β-amyloid (Aβ) found in senile plaques. The deleterious effects of Aβ deposition during the symptomatic stages of AD are undeniable, but the role of Aβ in earlier phases of the disease is still debated. During these early stages, AD cells exhibit alterations in numerous metabolic processes, such as perturbed mitochondrial function and loss of lipid homeostasis. These metabolic alterations occur early in AD, before the appearance of plaques, which has raised the possibility that other aspects of APP cleavage may be contributing to these metabolic changes. In this regard, increased levels of the C99 fragment have also been shown to contribute to AD pathogenesis.
The cleavage of C99 by γ-secretase activity occurs in specific areas of the ER called mitochondria-associated endoplasmic reticulum (ER) membranes (MAM). MAM is a lipid raft-like domain of the ER that regulates key cellular metabolic functions, such as lipid homeostasis and mitochondrial behavior. In a previous report, Drs. Estela Area Gomez and Eric Schon showed that ER-mitochondrial connectivity and MAM function are upregulated in AD.
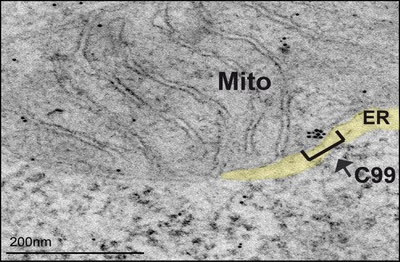
Figure 1: Representative immunoelectron microscopy image of PS-DKO cells incubated with antibodies against APP-CTF conjugated with immunogold particles, showing retention of C99 in MAM areas of the ER.
In the present study, published in The EMBO Journal, they show that the γ-secretase substrate C99, in addition to its endosomal localization, is also present in MAM domains. Thus, both the γ-secretase enzyme activity (i.e. presenilins) and its direct substrate (i.e. C99) are located in the same compartment, where the former can cleave the latter. Moreover, chemical and genetic alterations of γ-secretase activity provoke a significant increase in the amount of this APP processing fragment in MAM regions. The increased presence of C99 in MAM causes the upregulation of MAM functionality (as measured by cholesterol esterification activity) and greater apposition between ER and mitochondria. In addition, the higher concentration of MAM-localized C99 induces the recruitment of sphingomyelinase to this ER domain and the subsequent deregulation of sphingolipid homeostasis, followed by higher levels of ceramide on mitochondria that impair respiratory chain activity.
These results support a model in which, in addition to Aβ, a critical component of AD pathogenesis is mediated by C99 toxicity through its effects on MAM and mitochondria.
Estela Area Gomez, PhD
Assistant Professor in the Department of Neurology
eag2118@cumc.columbia.edu
Eric A. Schon, PhD
Lewis P. Rowland Professor of Neurology (in Genetics and Development)
eas3@cumc.columbia.edu
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.