
Columbia University
Irving Medical Center
Neurological Institute
710 West 168th Street, 3rd floor
(212) 305-1818
Featured Research
IN THE LAB:
Francesca Bartolini, PhD
 Francesca Bartolini, PhD
Francesca Bartolini, PhDA broadly accepted hypothesis states that oligomeric amyloid beta (Aβ) plays a crucial neurotoxic and synaptotoxic role in Alzheimer's disease (AD), and that hyperphosphorylated tau mediates or facilitates Aβ toxicity in dendrites and axons. The nature of the link between Aβ and tau remains, however, a subject of great debate. Research in our laboratory suggests that modifications of cytoskeletal dynamics by Aβ may be crucial to induce pathogenic changes in tau behavior and neuronal function.
Microtubule (MT) dynamics and tubulin post-translational modifications (PTMs) play critical roles in normal neuronal activity. Neurons possess extensive tubulin PTMs associated with MT longevity, and analyses of mammalian brain and cultured CNS neurons demonstrate enrichment of detyrosinated, acetylated, poly-glutamylated and Δ2-tubulin subunits. These PTMs are implicated in the regulation of MAPs, including tau itself, MT severing enzymes and binding to motors. Synaptic activation also appears to locally regulate PTMs associated with MT stability and dendritic spine maintenance depends on dynamic MTs. Thus, abnormal localized increases in MT stability in neurons have the potential to 1) impair MT-dependent axonal and dendritic trafficking; 2) disrupt synaptic transmission leading to spine collapse; 3) interfere with MT binding affinity of MT severing enzymes and MAPs including tau protein.
My laboratory studies the role of MT stability and its regulation in AD. We are primarily investigating roles for formins, a class of actin and MT stability regulators that act downstream of Rho GTPase activation. We found that Aβ induces the formation of a subset of stable detyrosinated MTs through RhoA and formin activities. We are now examining whether induction of this subset of stable MTs in neurons, in contrast to MT destabilization that occurs at a later stage, is a primary cause of the loss of synaptic activity and neurotoxicity caused by Aβ.
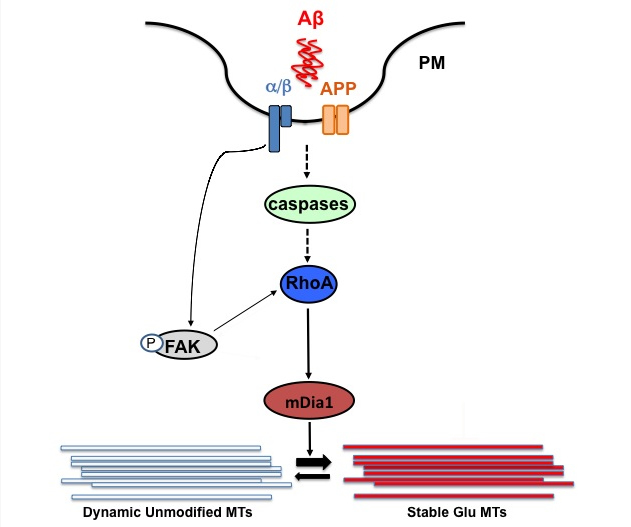 Proposed model for the pathway involved in Aβ induction of stable Glu MTs by Aβ in NIH3T3 cells. Oligomeric Aβ1-42 induces the formation of stable Glu MTs through a pathway regulated by integrin signaling, APP and Rho-mediated activation of mDia1. Signaling of Aβ through APP and integrins at the cell surface or in an endocytic compartment is likely to be the first step in the cascade that leads to RhoA-GTP and mDia1 activation through caspase activity. Active FAK and elevated RhoA-GTP levels are also potentially responsible for further FAK activation, which would then lead to focal adhesion assembly and formation of stable Glu MTs (red bars) from dynamic ones (blue bars) through the action of the formin mDia1. PM, plasma membrane.
|
Given the close association of synapse dysfunction/loss with the onset of neuropathological disorders such as AD and Parkinson's disease, we are also testing whether induction of MT stability is a general property of other amyloid proteins and an early cause of dysfunctional organelle trafficking and degradation, key cellular functions that go awry in progressive neurodegenerative disorders.
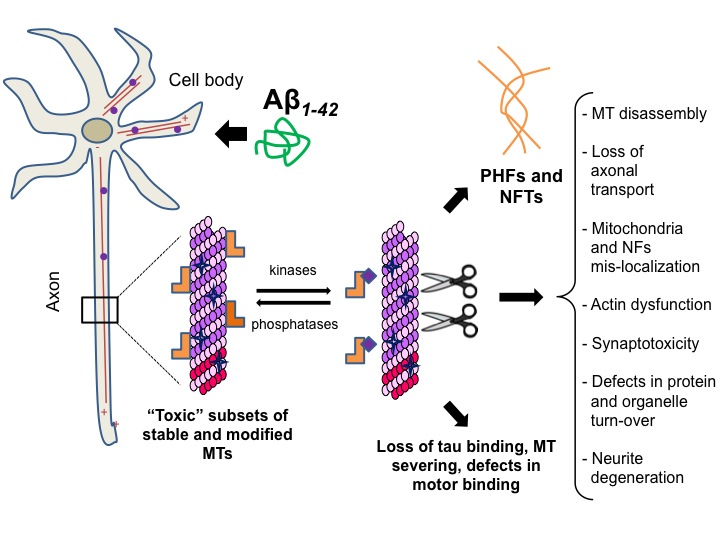 Our "Aβ to tau axis" model and loss of neuronal functions. Oligomeric Aβ induces the formation of 'toxic' subsets of modified stable MTs through receptor-mediated pathways in neurons. Abnormal accumulation of tubulin PTMs on stable MTs causes a cellular stress response that leads to activation of MT severing enzymes and accumulation of hyperphosphorylated tau in the attempt to restore normal levels of dynamic and unmodified MTs. Sustained induction of these neurotoxic pathways has the potential to induce loss of neuronal functions and death through the induction of MT disassembly, synaptotoxicity, defects in axonal transport and organelle clearance.
|
To tackle these questions, we employ biochemical and cell-biological approaches using immortalized non-neuronal cells, primary neuronal cultures and animal models of disease. Our work aims to shed light on the molecular nature of the link between amyloids and cytoskeletal changes and provide novel targets for therapeutic strategies to rescue impairment of cell function and cognition in neurodegenerative disease.
 Members of the Bartolini Laboratory, from left to right, Frank Yuan, Xiaoyi Qu, Mathilde Gagliardini, and Francesca Bartolini (PI).
|
Current projects in our lab include:
Xiaoyi Qu, a 2nd year PhD candidate student in the Department of Pathology and Cell Biology, is testing our hypothesis that abnormal activation of subsets of modified stable MTs by Aβ significantly contributes to tau hyperphosphorylation and synaptic dysfunction. We are also examining whether local induction of modified stable MTs alone is sufficient to decrease tau association rates with MTs, increase hyperphosphorylated tau levels, and affect dendritic spine morphology and density. This project may provide an explanation for the induction of synaptoxicity by Aβ through tau due to early changes in MT dynamics and tubulin PTMs.
Frank Yuan is a research assistant who is examining a previously unexplored role for members of the formin family of Rho GTPase effectors in the induction of synaptotoxicity by Aβ through the regulation of MT stability and tau hyperphosphorylation. This project, based on our published data, stems from collaborations with Taub members Drs. Shelanski and Lefort and aims at testing whether inhibition of formin activity confers protection against synaptotoxic damage and tau hyperphosphorylation induced by Aβ in isolated neurons and animal models of AD.
Mathilde Gagliardini is a visiting graduate student who is examining the distribution of subsets of modified tubulins and formin expression in animal models of AD and in post-mortem tissue isolated from AD patients at various degree of cognitive decline and disease progression.
Contact Us
630 West 168th Street
P&S Box 16
New York, NY 10032
Phone: 212 305-1818
Fax: 212 342-2849
Email: taubinstitute@columbia.edu
Brain Donation
Our ability to understand Alzheimer's disease is dependent upon studying brain tissue.
For more information, please contact Donovan Laing at (212) 305-9086 dal2190@cumc.columbia.edu
This website uses cookies as well as similar tools and technologies to understand visitors' experiences. By continuing to use this website, you consent to Columbia University's usage of cookies and similar technologies, in accordance with the Columbia University Website Cookie Notice.